












【佳学基因检测】基因检测找到一个听力障碍家庭的突变:ATP6V1B2和TJP2基因突变的病例分析
引言
基因检测作为现代医学的重要工具,在遗传病的诊断、治疗和预防中发挥了越来越重要的作用。本文将通过对一个特定家族的基因检测案例进行深入分析,介绍如何通过基因检测找到听力障碍家庭的基因突变,本文以ATP6V1B2和TJP2基因的变异做为病例。我们将详细介绍病例的临床表现、基因检测结果及其临床意义,以期为遗传性听力障碍的研究提供有价值的参考。
家族背景与临床表现
病例介绍
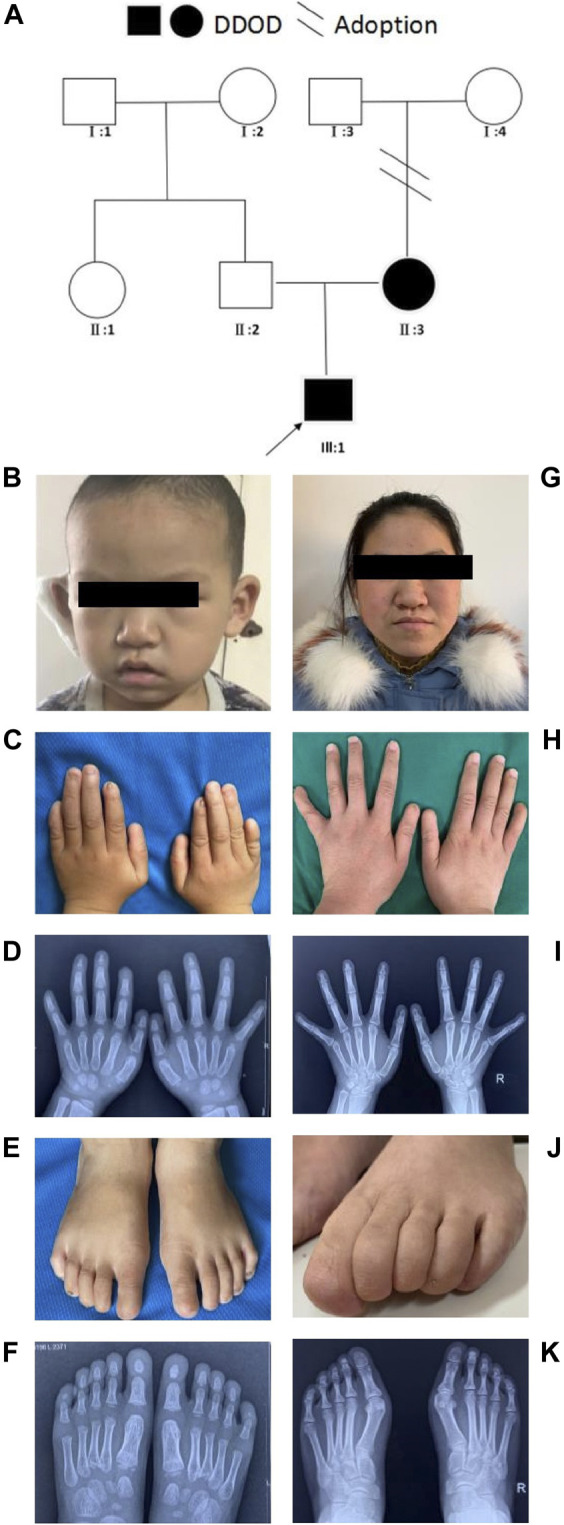
在研究的第一家族中,一名6岁的男孩因先天性听力损失于20个月大时首次就诊。经过单侧人工耳蜗植入手术后,他的听力和语言能力恢复正常。身体检查显示,他的双手和双脚均有指甲发育异常,包括第一和第五个手指缺少指甲,食指出现发育不全,而其余手指的指甲表面粗糙不平。他的拇指呈指状,但并非三关节拇指,且第五个手指明显短小。X光检查显示,食指的远端指骨缺失。
在他的双脚中,右脚的第一和第二趾缺少指甲,第三至第五趾的指甲发育不良;左脚的第一至第三趾缺少指甲,而第四和第五趾则有发育不全的指甲。足部检查及X光显示,第五个趾的远端指骨也存在发育不全。听力评估结果表明,他存在双侧重度感音神经性听力损失。
同样,男孩的母亲在20岁时也表现出相似的特征,包括双侧先天性感音神经性听力损失和指甲发育异常。她的趾甲发育受到严重影响,完全没有趾甲。母亲未接受听力干预,主要通过手语进行交流。
家族树
该家族的家族树显示出明显的遗传模式,母亲和儿子均表现出相似的听力和指甲异常症状,提示可能存在共同的遗传因素。
基因检测与突变分析
ATP6V1B2基因突变的鉴定
通过对家族成员进行三代全外显子测序(trio-WES),研究人员在ATP6V1B2基因中发现了一个已知的致病突变(c.1516C>T; p.R506*)。这一突变在多个报道中被认为与听力损失和指甲发育不全相关联,且与DOORS综合症(听力损失、指甲发育不全、骨发育不全、智力低下和癫痫)也存在相关性。
根据ACMG/AMP的分类标准,该突变被归类为致病性。其分类依据包括:
- PVS1:基于以往功能研究的证据。
- PP3:多种算法预测其为有害突变。
- PP5:在ClinVar和DVD数据库中被认为是致病性。
- PP1:家族成员间的共分离现象。
TJP2基因的突变分析
进一步的分析显示,母亲和儿子还携带TJP2基因中的一个变异(c.1590T>G; p.D530E),该基因被认为与肝内胆汁淤积和感音神经性听力损失相关。通过多种算法预测,该突变可能致病,并且在gnomAD数据库中未见记录。
此外,研究发现该变异可能影响mRNA的剪接,导致产生混合的PCR产物,进而提示突变可能对基因表达产生影响。根据ACMG/AMP的分类标准,该突变被归类为不确定意义变异(VUS),需要进一步的临床随访以评估其潜在影响。
临床意义与讨论
本研究的结果表明,ATP6V1B2和TJP2基因突变在家族性听力障碍中具有重要意义。ATP6V1B2的突变与听力损失和指甲发育不全的直接关联,为临床诊断提供了有力支持。同时,TJP2的变异可能为未来的研究方向提供新的思路。
在临床应用方面,基因检测可以帮助确诊遗传性听力障碍患者,进而为患者提供个性化的干预措施。此外,了解潜在的基因变异有助于对家族中其他成员的筛查与评估,降低疾病发作的风险。
听力基因检测结论
基因检测在遗传性疾病的诊断中展现了巨大的潜力。通过对特定家族的案例分析,我们揭示了与听力障碍相关的ATP6V1B2和TJP2基因的突变。这一研究为未来遗传性听力障碍的早期诊断和干预提供了宝贵的经验与数据支持,同时也提示了需要对不确定意义变异进行长期的临床观察与研究。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
