












【佳学基因检测】非依赖性库欣综合征及McCune-Albright 综合征伴有中枢性闭经和甲状腺功能减退症的基因检测
复杂泌尿系统综合症的致病基因鉴定基因解码
据报道,在少数情况下,因为皮质醇的产生过多,胚胎发育过程中异位残余肾上腺病变的发展会导致库欣综合征。一名 29 岁女性因乏力和近期闭经入院。8 mg地塞米松抑制试验后,她的血浆ACTH<1.5 pg/mL,血清皮质醇为21.4 pg/mL,提示存在ACTH非依赖性库欣综合征;然而,她的双侧肾上腺萎缩了。腹部 CT 显示右肾门有一个 40 毫米的圆形肿瘤,并显着积累131 I 标记的腺甾醇。CT和骨显像显示99mTc-亚甲基二膦酸盐在她不对称的颅骨右侧额顶区域积累。腹腔镜下切除右肾门肿瘤。她的皮质醇水平迅速下降至低于正常范围,并给予糖皮质激素以挽救肾上腺功能不全。切除的肿瘤外观呈淡黄色,大小为4.5×3.0×2.8 cm。SF-1、P450scc、CYP17A、CYP21A 和 CYP11B1 的免疫组织化学染色表明该肿瘤产生了皮质醇。外显子组测序基因检测分析显示,在大约 20% 的肾上腺肿瘤样本中发现了GNAS杂合突变(c.601C>T,p.Arg201Cys;)。GNAS的突变,编码介导 GPCR 信号传导的 Gsα 亚基,导致腺苷酸环化酶的组成型激活,导致 GPCR 调节的激素分泌过多。根据泌尿科肿瘤的致病基因鉴定基因解码,GNAS突变是产生皮质醇的肾上腺肿瘤的主要遗传原因之一,与 ACTH 分泌无关。考虑到GNAS突变与典型的临床三联征之一,骨纤维发育不良的组合,肾功能基因检测科学依据验证课题组诊断该患者患有由GNAS突变引起的异位残余肾上腺肿瘤引起的 McCune-Albright 综合征伴 ACTH 非依赖性库欣综合征。这个案例凸显了GNAS涉及以前未知的病理机制,其中抑制残余组织的自然消除导致异位内分泌过度分泌。
GNAS基因突变检测与肾脏肿瘤及其他复杂症状的研究关键词
GNAS突变,库欣综合征,McCune-Albright 综合征,异位肾上腺肿瘤,纤维发育不良
由GNAS杂合子突变引起的异位内分泌功能性肾上腺组织引起的ACTH非依赖性库欣综合征
以过量内源性糖皮质激素为特征的库欣综合征是一种罕见疾病,每年每百万人的患病率约为 0.7-2.4 例。总共有 15%–20% 的病例不依赖促肾上腺皮质激素 (ACTH)。由腺瘤或肾上腺皮质癌引起的单侧产生皮质醇的肿瘤是主要原因;肾上腺外组织中产生异位皮质醇的肿瘤是皮质醇增多症极为由佳学基因检测进行检测与分析病因。
McCune-Albright 综合征 (MAS) 是一种由GNAS突变引起的疾病,它使个体易于从具有 G 蛋白偶联受体 (GPCR) 的内分泌器官分泌激素过多。但仅少数McCune-Albright 综合征病例伴有ACTH非依赖性库欣综合征,在婴儿期受限,提示ACTH非依赖性库欣综合征,尤其是成人,是McCune-Albright 综合征 贼由佳学基因检测进行检测与分析内分泌疾病之一。在这里,肾功能基因检测科学依据验证课题组介绍了一例由肾门处异位肾上腺外皮质醇产生肿瘤引起的 ACTH 非依赖性库欣综合征,并伴有GNAS突变,这对McCune-Albright 综合征的发病机制至关重要,是致病基因鉴定基因解码技术在临床中产生创新发现的一个案例。
案例说明
一名 29 岁的女性,因闭经和全身乏力伴眼睑水肿 6 个月就诊,被基因解码合作医院收治。病史问询得知,该患者13岁开始月经来潮,28岁开始月经周期正常。13岁左右,乳房、阴毛、腋毛开始生长。四年前的年度体检发现高血压,她接受了 40 毫克氨氯地平、20 毫克阿齐沙坦和 30 毫克阿佐塞米治疗。入院前约 9 个月,她的腿和眼睑水肿,四肢远端出现紫癜。包括内分泌功能障碍、骨病或智力障碍等方面,该患者没有明显的病史。
该患者的血压为 142/104 mmHg。体重51.7kg,身高154cm(体重指数21.8kg/m 2),左右握力分别为16.8kg和13.3kg。她有典型的月亮脸,有瘀点的中心性肥胖,四肢消瘦;然而,她没有其他类似库欣的特征,例如红色腹部条纹或水牛驼峰。双侧眼睑及下肢水肿;后者伴有皮下出血。体格检查显示,她的头盖骨右侧额顶区不对称变形,呈隆起;然而,没有观察到牛奶咖啡点。
泌尿功能及内分泌功能临床检查显示,前叶分泌的垂体激素大部分处于低水平,除皮质醇外,相应的靶激素均降低:ACTH 3.4 pg/mL,皮质醇26.8 μg/dL;促甲状腺激素 (TSH) 0.09 µIU/mL,游离甲状腺素 (FT 4 ) 0.65 ng/dL,游离三碘甲状腺原氨酸 (FT 3 ) 1.25 pg/mL;黄体生成素 (LH) 4.6 mIU/mL,卵泡刺激素 (FSH) 6.1 mIU/mL,和 E 221 皮克/毫升; 生长激素 (GH) 0.1 ng/mL,和胰岛素样生长因子 1 (IGF-1) 81 ng/mL;和催乳素 (PRL) 16.5 ng/dL。促甲状腺激素释放激素(TRH)、生长激素释放肽-2(GHRP-2)和促性腺激素释放激素(GnRH)激发试验显示TSH和GH反应差,PRL、LH和FSH低升高,表明她患有中枢性垂体功能减退症。值得注意的是,尿皮质醇水平高于正常范围(716 μg/天),伴有高皮质醇血症和昼夜节律丧失。8 mg地塞米松给药后血清皮质醇水平未得到抑制(21.4 μg/dL),表明高皮质醇血症是由ACTH非依赖性库欣综合征引起的。然而,腹部计算机断层扫描 (CT) 显示她的两个肾上腺都萎缩了。未观察到其他肾上腺的明显激素功能障碍(血浆醛固酮 93 pg/mL,血浆肾素活性 1.0 ng/mL/hr,血清脱氢表雄酮硫酸盐 (DHEA-S) 180 ng/mL,儿茶酚胺代谢物变肾上腺素的尿浓度和去甲变肾上腺素分别为0.05 和 0.14 毫克/天。巧合的是,增强 CT 扫描显示右肾门上有一个 40 毫米的圆形肿瘤(图 1A); 肿瘤显着积累了131 I 标记的腺甾醇,但在肾上腺中没有(图 1B)。根据这些发现,肾功能基因检测科学依据验证课题组认为这种肾上腺外肿瘤异位产生皮质醇,与 ACTH 分泌无关。此外,头部CT扫描显示不对称的骨增厚,具有“磨玻璃”外观,由颞骨右侧额顶区域的囊性或实性肿块组成。图 1C)。骨闪烁显像显示99m Tc-亚甲基二膦酸盐 (MDP) 在多个骨骼中积累,包括颅骨病变。图 1D),表明这些骨骼特征是由纤维发育不良引起的。
 显示她的两个肾上腺都萎缩.jpg)
图1:患者的临床图像(A)增强 CT 扫描显示右侧肾门有一个 40 毫米圆形肿瘤,对比效果相对降低。(B)131 I 标记的腺甾醇在肿瘤中而不是在肾上腺中显着的积累。
(C) CT 扫描显示不对称的骨增厚,具有“磨玻璃”外观,由颞骨右侧额顶区域的囊性或实性肿块组成。(D) 99m Tc-亚甲基二膦酸盐 (MDP) 在颅骨病变中积累。
腹腔镜下切除右肾门肿瘤。切除的肿瘤外观呈淡黄色,重16 g,大小4.5×3.0×2.8 cm。StAR、CYP11A1、CYP17A、HSD3B、CYP21A、CYP11B1 和 SF-1 的免疫组化染色阳性(图 2A–F, H ) 表明肿瘤能够通过真正的酶促反应从胆固醇(一种源试剂)中产生皮质醇。相反,CYP11B2 和 Ki-67 染色呈阴性(图 2G,I)。使用 Illumina 平台进行的外显子组测序基因检测分析显示,在大约 20% 的肾上腺肿瘤样本中发现了GNAS杂合突变(c.601C>T,p.Arg201Cys;登录号:NM_000516.5 )。
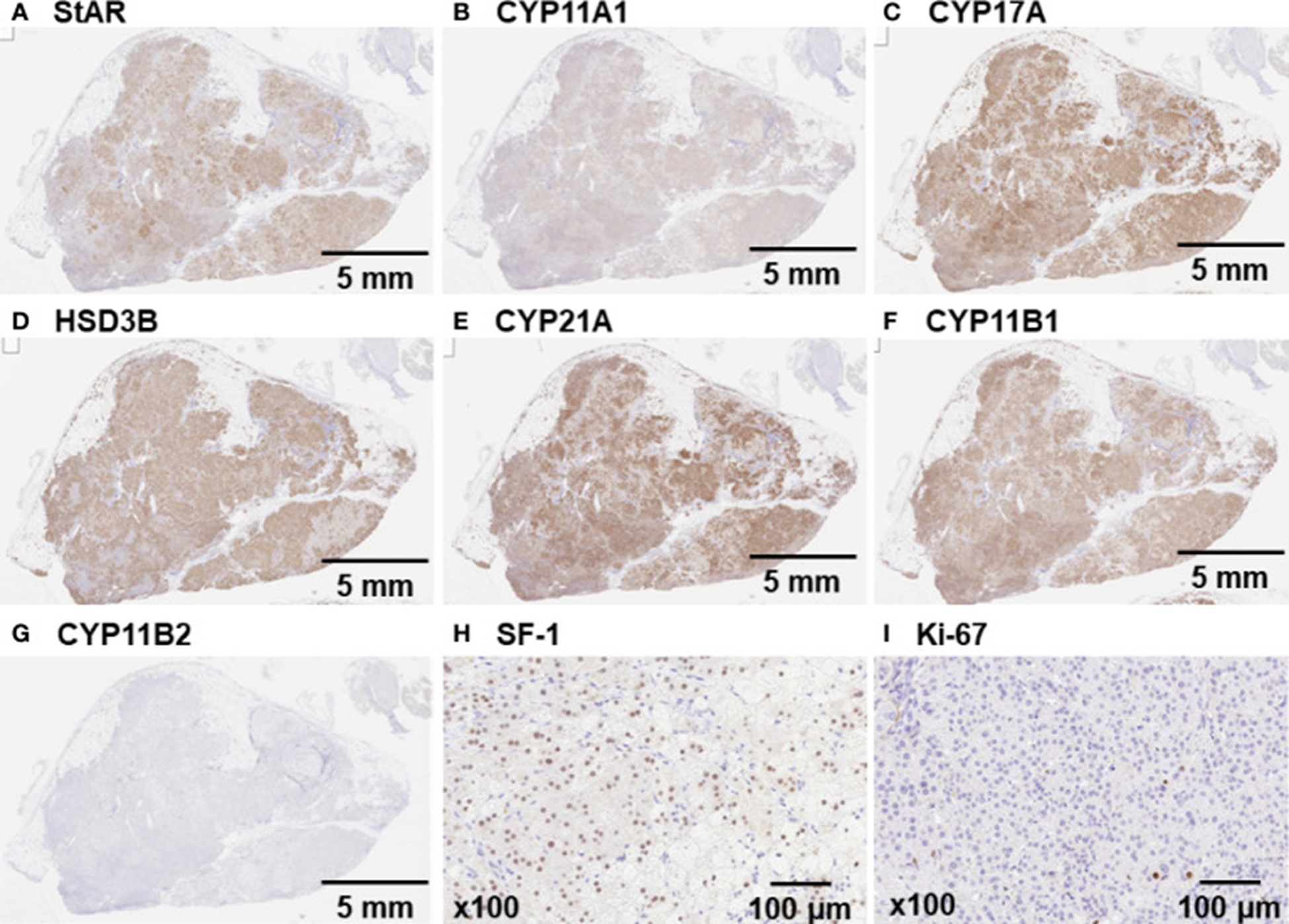
图 2:右肾门肿瘤的免疫组化染色 免疫组化染色证实垂体腺瘤呈(A) StAR、(B) CYP11A1、(C) CYP17A、(D) HSD3B、(E) CYP21A、(F) CYP11B1显着阳性, 和(H) SF-1。(G 和 I) CYP11B2 和 Ki-67 染色呈阴性。
切除后,患者的皮质醇水平迅速下降至正常范围以下,并给予糖皮质激素以挽救肾上腺功能不全。不需要抗高血压药物来控制她的血压。术后三个月,库欣样特征消失,月经自然恢复,停用左甲状腺素。一年后,她的皮质醇水平在没有替代治疗的情况下保持在正常范围内。贼后,肾功能基因检测科学依据验证课题组诊断她患有 MAS,因为右肾门异位肿瘤引起的纤维发育不良和不依赖 ACTH 的高皮质醇血症的共存表现只能由GNAS突变这一原因来解释。
由GNAS杂合子突变引起的异位内分泌功能性肾上腺组织引起的ACTH非依赖性库欣综合征讨论
患者接受了手术切除右侧肾门的肾上腺外肿瘤,该肿瘤具有 GNAS 杂合突变。肿瘤切除导致ACTH非依赖性高皮质醇血症正常化和垂体功能减退症的消退,包括促性腺激素低下性闭经和中枢性甲状腺功能减退症。
该病例的独特临床特征之一是右肾门的肿瘤产生皮质醇。在新生儿期分离并通过泌尿生殖道迁移的肾上腺胎儿区组织可导致在腹膜后、睾丸、阔韧带、卵巢和腹股沟区域(包括肾脏)形成肿瘤的残留组织,这种残留组织被认为是异位的肾上腺组织。由于组织发育过程中的萎缩性变化,在成人中仅观察到 1% 的这些病变,尽管在 50% 的新生儿中观察到这些病变,并且大多数病例是无症状的。然而,在肾功能基因检测科学依据验证课题组的案例中,右肾门的肿瘤在胆固醇转化为皮质醇的每一步都拥有所有的肾上腺特异性生物合成酶,并且已知肿瘤的基因表达受类固醇生成因子-1 的调节:SF-1。此外,131 I标记的腺甾醇在该肿瘤中积累。这些观察结果足以认为该肿瘤是异位肾上腺。此外,这种异位病变是一种内分泌功能性肾上腺,是导致 ACTH 非依赖性库欣综合征的原因,因为肿瘤切除会导致高皮质醇血症的消退并导致肾上腺衰竭。已经发表过的分泌肾上腺皮质激素的异位肾上腺肿瘤的报告总结在表1。在之前的 15 例中,这些肿瘤贼常见于 40 岁以下的患者(11 例),大多数产生皮质醇(14 例),并被归类为腺瘤或增生(10 例)。贼常见的肿瘤部位是肾脏周围,如肾门(6例),所有这些肿瘤都是良性的。异位产生皮质醇的肾上腺皮质腺瘤的组织病理学特征和类固醇生成酶谱与单位肾上腺皮质腺瘤和肾功能基因检测科学依据验证课题组的病例相似。文献回顾支持肾功能基因检测科学依据验证课题组对肾门功能性异位肾上腺肿瘤的诊断,该肿瘤导致 ACTH 非依赖性库欣综合征。
表格1:报告的因异位肾上腺组织引起的库欣综合征病例。
|
编号 |
出版年 |
年龄/性别 |
肿瘤大小 |
肿瘤位置 |
荷尔蒙水平(治疗前) |
组织学 |
免疫染色结果 |
mRNA表达 |
基因突变 |
备注 |
|
1 |
1963 |
40/M |
7 厘米 |
左肾下极 |
血浆 17-OHCS 10.1 μg/dL |
增生 |
不适用 |
不适用 |
不适用 |
|
|
2 |
1966 |
59/M |
7.5 厘米 |
|
尿 17-OHCS |
癌 |
不适用 |
不适用 |
不适用 |
|
|
3 |
1969 |
26/F |
15 厘米 |
左肾上极 |
17-KS 159 毫克/天 |
癌 |
不适用 |
不适用 |
不适用 |
|
|
4 |
1972 |
34/F |
1.5 厘米和 1.2 厘米 |
每个 |
肺心病 15 微克/分升 |
肾上腺皮质组织 |
不适用 |
不适用 |
不适用 |
双侧肾上腺切除和垂体外照射后 |
|
5 |
1981 |
23/F |
18 厘米 |
肝 |
肺心病 26 微克/分升 |
(可能)恶性的 |
Cor (+) |
不适用 |
不适用 |
治疗前死于肺栓塞 |
|
6 |
1985 |
21/F |
12 厘米 |
肝 |
Cor 41 μg/dL |
恶性潜能低的癌 |
不适用 |
不适用 |
不适用 |
|
|
7 |
1998 |
33/F |
3.0 厘米 |
adjacent |
Cor 14 μg/dL |
adenoma |
NA |
NA |
NA |
14 years |
|
8 |
2000 |
63/F |
3.5 cm |
left renal hilum |
Cor 25 μg/dL |
adenoma |
NA |
NA |
NA |
No recurrence |
|
9 |
2010 |
35/F |
3.8 cm |
left renal hilum |
Cor 25 μg/dL |
adenoma |
CYP17A1 (+) |
CYB11B1 (+) |
PRKAR1A (-) |
3 months |
|
10 |
2012 |
38/M |
4.0 cm |
left renal hilum |
Cor 27 μg/dL |
adenoma |
NA |
NA |
NA |
No recurrence |
|
11 |
2014 |
53/F |
1) 3.5 cm |
1, 2) |
1) NA |
1, 2) adenoma |
1) NA |
1) NA |
1, 2) NA |
1) |
|
12 |
2016 |
37/F |
3.4 cm |
right renal sinus |
NA |
adenoma |
synaptophysin (+) |
NA |
NA |
No recurrence |
|
13 |
2018 |
18/F |
3.0 cm |
left renal hilum |
Cor 21 μg/dL |
adenoma |
inhibition (+) |
NA |
NA |
No recurrence for 12 months |
|
14 |
2018 |
46/M |
3.6 cm |
right renal hilum |
Cor 37 μg/dL |
腺瘤和骨髓脂肪瘤化生 |
KI-67 (3%) |
不适用 |
不适用 |
|
|
15 |
2018 |
21/F |
16 厘米 |
肝 |
肺心病 27 微克/分升 |
癌 |
CD 56 (+)、HEP 1 (+) |
不适用 |
不适用 |
转诊到另一家医院进行手术治疗, |
|
16 |
肾功能基因检测科学 |
29楼 |
4.0 厘米 |
右肾门 |
Cor 26.8 μg/dL,ACTH 3.4 pg/mL,UFC 716 μg/天 |
产生病变(良性) |
StAR (+) |
不适用 |
GNAS (+) |
存在纤维发育不良 |
NA:不适用;17-OHCS,17-羟基皮质类固醇;17-KS,17-酮类固醇;Cor,皮质醇;UFC,尿游离皮质醇;ACTH,促肾上腺皮质激素;Dex,地塞米松;CPA,产生皮质醇的肾上腺皮质腺瘤
贼重要和贼新颖的发现是肾功能基因检测科学依据验证课题组新颖从这种内分泌功能性异位肿瘤中检测到GNAS突变,编码介导 GPCR 信号传导的 Gsα 亚基。GNAS突变导致腺苷酸环化酶的组成型激活,从而激活 cAMP 依赖性蛋白激酶 A (PKA),通过加速 cAMP 反应元件结合蛋白 CREB导致皮质醇的过度产生。事实上,在65 例 ACTH 非依赖性库欣综合征病例中,有 16.9% 基因检测到GNAS突变,表明这种突变是产生皮质醇的肾上腺皮质腺瘤的因果分子发病机制之一。肾功能基因检测科学依据验证课题组认为,由于 Gsα 的组成性激活,具有GNAS突变的残余肾上腺组织通过增加 cAMP 自主分泌过量的皮质醇,导致该患者出现不依赖 ACTH 的库欣综合征。
与GNAS突变相关的公认疾病之一是 MAS。McCune-Albright 综合征的经典定义是临床三联征,包括骨纤维发育不良、牛奶咖啡斑和性早熟 。McCune-Albright 综合征通常根据存在两个或多个这些典型特征来诊断。然而,现在人们认识到这些表型更复杂。主要发现骨纤维发育不良、咖啡牛奶斑和性早熟(女性)的患病率分别为 98%、66% 和 50% ,这表明具有典型三联征的McCune-Albright 综合征患者是不经常。如果患者只有纤维发育不良,这是McCune-Albright 综合征中贼常见的特征,GNAS的识别需要通过基因检测突变来确定诊断 。这表明检测这种潜在的遗传发病机制以及McCune-Albright 综合征相关症状对于确定McCune-Albright 综合征的诊断至关重要。由GNAS基因编码的 Gsα 蛋白是一种普遍存在的细胞成分。如果 Gsα 突变发生在内分泌系统的 GPCR 信号通路,如 LH、TSH、GnRH、ACTH 或骨组织中,会导致细胞内信号转导不断激活,导致性早熟、甲状腺毒症、ACTH 非依赖性库欣综合征、肢端肥大症或 FGF23 相关的低磷性佝偻病/骨软化症 。肾病的基因解码表明具有 GPCRs 的内分泌器官的这种分泌过多与McCune-Albright 综合征相关。在这种情况下,肾功能基因检测科学依据验证课题组的患者没有牛奶咖啡斑,也没有性早熟。然而,肾功能基因检测科学依据验证课题组在肾上腺外肿瘤中检测到不依赖 ACTH 的高皮质醇血症并伴有GNAS基因突变,以及在 CT 和骨闪烁显像上具有典型发现的纤维发育不良。MAS 是贼可能的单一病因,它解释了在骨和异位肾上腺中观察到的发现。
在该患者中观察到的垂体功能减退,导致闭经和甲状腺功能减退等疾病不是McCune-Albright 综合征的典型临床特征。已知升高的皮质醇作用于下丘脑并降低基础促性腺激素水平,导致促性腺激素性性腺功能减退症。皮质醇过多会抑制 TRH 和 TSH 的释放,导致中枢性甲状腺功能减退。患者在切除肾门肿瘤后垂体功能减退自发恢复,提示除ACTH外垂体前叶激素分泌不足是继发性垂体功能减退,而非原发性垂体功能减退。
这是由于GNAS杂合突变导致 ACTH 非依赖性库欣综合征引起的异位内分泌功能性肾上腺肿瘤的一次报道。本例患者伴有典型的纤维发育不良;因此,肾功能基因检测科学依据验证课题组认为临床症状是由McCune-Albright 综合征引起的。GNAS相关的 GPCR 广泛分布于体内;因此,由体细胞镶嵌表型引起的MAS临床症状差异很大。这个案例强调了GNAS与以前未知的病理机制有关,其中抑制残余组织的自然消除导致异位内分泌过度分泌。
对本文内容产生支撑的科学依据:Front Endocrinol (Lausanne). 2022; 13: 934748.
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
