












【佳学基因检测】常染色体显性遗传4型脊髓小脑共济失调的诊断基因检测
脊髓小脑共济失调4型是常色体隐性遗传还是常染色体显性遗传?
SCA4 具有常染色体显性遗传方式。大多数被诊断为 SCA4 的个体有一位受累的父母。由于 SCA4 中存在遗传预期现象(anticipation),受累的父母通常疾病较轻,发病年龄比其受累的子女晚。每位 SCA4 患者的子女有 50% 的几率遗传 ZFHX3 基因中的异常 GGC 重复扩增。一旦在受累家族成员中发现致病性 ZFHX3 GGC 重复扩增,就可以对有风险的家族成员进行预测性检测,并可开展产前或胚胎植入前的基因检测。
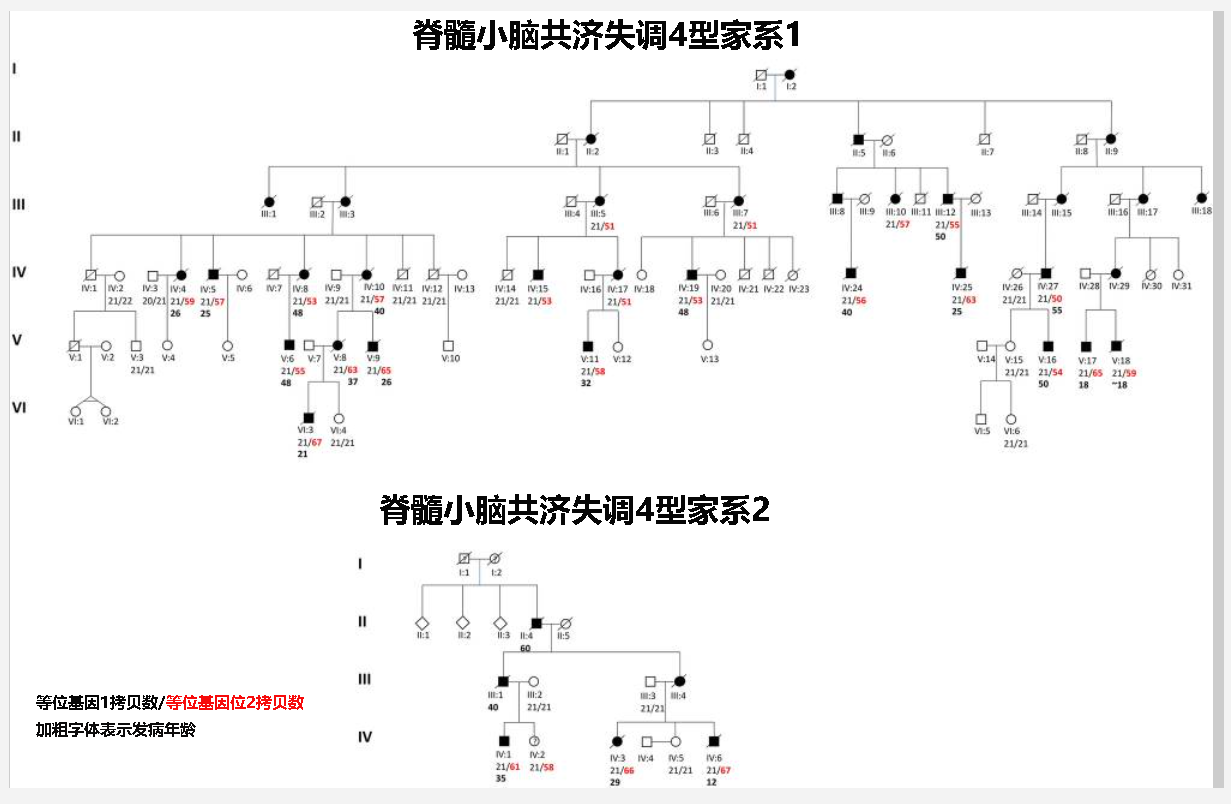
脊髓小脑共济失调4型病例的家族遗传谱
下图是佳学基因收集提供的一个脊髓小脑共济失调4型的家系遗传图。该图显示了两个家系的遗传情况。黑色实框显示为患病者,白色框显示为表现正常。罗马数字表示该家系中的代际编号。用斜杠表示的两个数字是ZFHX3 基因中的 GGC重复数目。红色数字是具有异常重复数的等位基因。从下图中的两个家系的遗传情况来看,男性和女性都会患病,表现为常染色体显性遗传。
常染色体显性遗传4型脊髓小脑共济失调(Spinocerebellar Ataxia, Autosomal Recessive 4)的诊断基因检测
常染色体显性遗传4型脊髓小脑共济失调(SCA4)是一种罕见的神经系统疾病,主要表现为运动协调障碍、平衡问题和言语困难。由于其遗传特性,基因检测在早期诊断和管理中起着至关重要的作用。 通过基因检测,可以准确识别与SCA4相关的基因突变,帮助患者及其家庭了解疾病的遗传风险。这种检测不仅有助于确诊,还能为患者提供个性化的治疗方案和预后评估。对于有家族史的个体,基因检测可以揭示其携带者状态,从而为生育计划提供科学依据,减少后代患病的风险。 此外,基因检测的结果可以为临床研究提供重要数据,推动新疗法的开发。随着基因组学的进步,越来越多的治疗策略正在被探索,包括基因治疗和靶向药物,这些都依赖于对特定基因突变的了解。 鼓励进行基因检测,不仅是对患者及其家庭的责任,也是对科学研究的支持。通过早期诊断和干预,可以显著改善患者的生活质量,减轻家庭的心理负担。因此,推广SCA4的基因检测,能够为患者带来希望,帮助他们更好地应对这一挑战。
常染色体显性遗传4型脊髓小脑共济失调(Spinocerebellar Ataxia, Autosomal Recessive 4)和遗传有关系吗?
常染色体显性遗传4型脊髓小脑共济失调(SCA4)与遗传有密切关系。SCA4是一种由特定基因突变引起的神经系统疾病,通常表现为运动协调障碍、平衡问题和其他神经功能障碍。该病的遗传模式为常染色体隐性,这意味着患者必须从每位父母那里继承一个突变基因才能表现出疾病症状。 鼓励基因检测的原因在于,早期识别携带者和患者可以帮助家庭做出知情的生育选择,并为潜在的患者提供早期干预和管理方案。通过基因检测,能够确认是否存在与SCA4相关的基因突变,从而为患者及其家属提供明确的遗传风险评估。 此外,基因检测还可以帮助医生制定个性化的治疗计划,尽早介入可能的康复措施,改善患者的生活质量。对于有家族史的人群,基因检测尤为重要,因为这可以揭示他们是否为携带者,进而影响其后代的健康风险。 总之,常染色体显性遗传4型脊髓小脑共济失调与遗传有直接关系,基因检测不仅能够提供重要的遗传信息,还能为患者及其家庭带来希望和支持。因此,鼓励进行基因检测是非常必要的。
常染色体显性遗传4型脊髓小脑共济失调(Spinocerebellar Ataxia, Autosomal Recessive 4)基因检测是个体化治疗决策的科学基础
常染色体显性遗传4型脊髓小脑共济失调(SCA4)是一种神经系统遗传疾病,患者通常表现为运动协调障碍、平衡失调等症状。基因检测在此类疾病的管理中具有重要意义,能够为个体化治疗决策提供科学依据。 首先,基因检测可以明确诊断。通过检测SCA4相关基因的突变,医生能够确认患者是否携带致病基因,从而避免误诊和不必要的治疗。这种精准的诊断为制定个体化的治疗方案奠定了基础。 其次,基因检测有助于评估疾病的遗传风险。了解患者的基因状态后,家庭成员可以进行相应的检测,评估他们的携带风险。这对于计划生育和早期干预具有重要意义,能够帮助家庭做出更明智的决策。 此外,基因检测还可以为临床试验和新疗法的选择提供指导。随着基因治疗和靶向治疗的发展,了解患者的具体基因突变类型可以帮助医生选择最合适的治疗方案,提高治疗效果。 最后,基因检测的结果可以促进患者及其家庭的心理支持。明确的基因检测结果能够减轻患者及其家属的焦虑,帮助他们更好地理解疾病,增强应对能力。 综上所述,常染色体显性遗传4型脊髓小脑共济失调的基因检测不仅是确诊的关键工具,更是个体化治疗决策的重要科学基础。通过基因检测,患者能够获得更精准的医疗服务,提高生活质量,增强对疾病的掌控感。
常染色体显性遗传4型脊髓小脑共济失调的临床表征
发病年龄
-
起病年龄范围广,从 12 岁到 65 岁。
-
约 10% 个体属于早发型(25 岁前发病)。
小脑/脑干累及表现(进行性)
-
步态共济失调
-
所有患者均表现为步态不稳和平衡障碍,通常是最早出现的症状。
-
-
肢体共济失调
-
可从症状起始即出现,也可能在疾病进展中才显现。
-
-
眼球运动异常
-
慢性或低幅度扫视(ocular saccades)、注视受限等几乎出现在所有患者。
-
其他可能表现:头眼滞后、扫视追踪障碍、扫视入侵、高幅扫视、少数患者可出现眼震或前庭-眼反射抑制障碍。
-
随疾病进展,水平或垂直注视范围可能受限。
-
-
构音障碍(Dysarthria)
-
近所有患者出现,小脑型构音不清、断续性语言(scanning speech)。
-
-
吞咽障碍(Dysphagia)
-
约 65% 患者随疾病进展出现,表现为口腔肌肉协调障碍及咽部残留。
-
感觉神经病变
-
罕见情况下,远端肢体感觉障碍可作为早期或首发症状。
-
随疾病进展,几乎所有患者出现感觉神经病变。
-
临床检查:振动觉下降普遍存在;部分患者触痛、温度觉、轻触觉和本体觉受损。
-
罗姆伯格试验(Romberg test)可阳性。
-
神经生理学检查:感觉神经动作电位振幅下降或消失,可早于平衡和步态障碍出现。
上、下运动神经元损害
-
上运动神经元(UMN):约 85% 患者可见足底伸肌反射阳性。
-
下运动神经元(LMN):30%–100% 患者可出现轻度肌无力、肌肉萎缩。
-
随疾病进展,腱反射多减弱或消失。
自主神经功能障碍
-
早期较少见,疾病进展中可明显,包括:
-
严重直立性低血压
-
尿急或尿失禁
-
便秘或腹泻
-
体重和肌肉状态
-
疾病晚期可能出现体重下降和/或肌肉萎缩。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
