












【佳学基因检测】肌营养不良基因检测复杂性的解析策略
佳学基因导读:
在 961 名临床怀疑患有杜氏肌营养不良症的患者中,有 105 名被诊断患有其他肌营养不良症 (OMD),其中最常见的是LGMD2E(变体SGCB c.544A>C)。在进行致病基因鉴定基因检测中,检测项目的选择和指定,往往需要选择拓展的检测范围 ,以便解决临床症状出现的复杂性,增加检测分析的准确性。
大多数其他肌营养不良症 (OMD)患者患有常染色体隐性疾病,包括肢带型肌营养不良症 (LGMD)、Bethlem、Ullrich 先天性肌病和 Emery-Driefuss 肌营养不良症。3.5% 的患者被确诊患有其他疾病,如夏科-马里图斯病和杆状体肌病。一小部分患者(0.6%)仍未确诊。在总共 78 个已识别的基因变异中,有 44 个是新的。有趣的是,三分之一的其他肌营养不良症 (OMD)患者被发现患有 LGMD2E/R4,这是一种严重的 LGMD,会使幼儿出现与 DMD 相似的临床症状。几乎三分之一的无关 LGMD2E/R4 患者在SGCB基因中具有相同的点突变(c.544A>C),提示存在奠基者效应,这是肌营养不良基因检测进行描述。
肌营养不良基因检测复杂性的解析策略关键词:
常染色体隐性遗传病、杜氏肌营养不良症、肢带型肌营养不良症 2E-R4、肌营养不良症、下一代测序、重叠症状
肌营养不良基因检测临床大数据分析中所有经分子学证实的 LGMD 都是临床上怀疑为 DMD/BMD 的。在肌营养不良基因检测临床大数据分析中,肌营养不良基因检测试图了解患者的发病年龄,这可能是区分 DMD/BMD 与其他肌营养不良症的一个区别因素。根据发病年龄,很明显,在肌营养不良基因检测的队列中,LGMD 2E 的严重程度与 DMD 最接近,其中变体 c.544A>C 最为常见,临床症状也表现出相同的症状。
为什么要进行肌营养不良基因检测的大数据比较分析
下一代 DNA 测序方法对临床疑似杜氏肌营养不良症 (DMD) 患者的精确和明确诊断做出了巨大贡献,从而提高了诊断率。DMD 是一种单基因、X 连锁隐性、退行性神经肌肉疾病,影响年轻男性,并导致致命的心肺和呼吸衰竭。及早准确识别肌营养不良蛋白 ( DMD ) 基因变异对于诊断该疾病至关重要,并正在成为有效疾病管理策略的重要组成部分。由于 DMD 是一种进行性疾病,因此需要尽早通过咨询启动护理指南和疾病预防策略,特别是因为现在已有针对一部分患者的有效疗法。
在最近的一项研究中,肌营养不良基因检测报告了961 名无亲缘关系、临床怀疑患有 DMD 的男性患者的DMD基因突变谱。肌营养不良基因检测采用了一种分子诊断方法,这种方法对大多数患者来说都是经济有效的,并遵循一个系统的过程,该过程依次包括使用 mPCR 识别热点缺失、使用 MLPA 识别大量缺失和重复以及使用 NGS 先天性肌营养不良症基因面板识别小插入/缺失和点突变。近 85% (816/961) 的患者被确诊为DMD基因变异。其余 15% 的患者 (145/961) 缺乏DMD基因变异,佳学基因检测对这一部分检测样本进行了深度分析,以提高解决临床需求的目的。
材料与方法
患者纳入和排除标准
2006 年至 2013 年间,佳学基因检测及其合作机构接收了 961 名临床怀疑患有 DMD/BMD 的男性患者,其中 99% 是无亲属关系的,进行分子诊断。这些患者大多是通过农村 MDCRC 社区遗传学计划以及来自合作的医院和诊所转诊到 MDCRC 的。
肌营养不良基因检测的 961 名患者队列包括年龄在 2-35 岁之间且表现出 DMD 特征症状的年轻男性。排除年龄在 35 岁以上的男性患者和具有 DMD 样症状的女性患者。在采集样本之前,已获得每位患者(或父母/监护人)的知情书面同意。
样本采集及 DNA 分离和保存
使用 EDTA 真空采血管从临床疑似 DMD 患者身上采集 3 毫升血液样本。使用之前描述的简单脱盐方案提取基因组 DNA,并储存在 -20°C 下直至进一步使用。
DNA 样本的多重 PCR、多重连接依赖性探针扩增 (MLPA) 和下一代测序
按照图 1中的算法对基因组 DNA 的分子诊断检查进行了分析。首先进行 mPCR,旨在检测DMD基因热点区域的任何缺失,该区域覆盖 30 个外显子,正如肌营养不良基因检测之前描述的。接下来在 mPCR 阴性样本中进行 MLPA 分析,使用商业购买的 PO34 和 PO35 探针。mPCR 或 MLPA 后没有发现DMD基因变异,因此使用下一代序列 (NGS) 分析对样本进行测序。生成的文库肌营养不良基因检测的 Illumina HiSeq 系列 4500 上进行测序,以 80-100 倍的靶向测序深度生成 2 × 150 bp 序列读数。仅在肌营养不良症和先天性肌病组基因中发现的非同义和剪接位点变体用于临床解释。使用 ABI 3730xl 仪器上的标准协议进行 Sanger 测序,以验证 NGS 识别的点突变的存在。
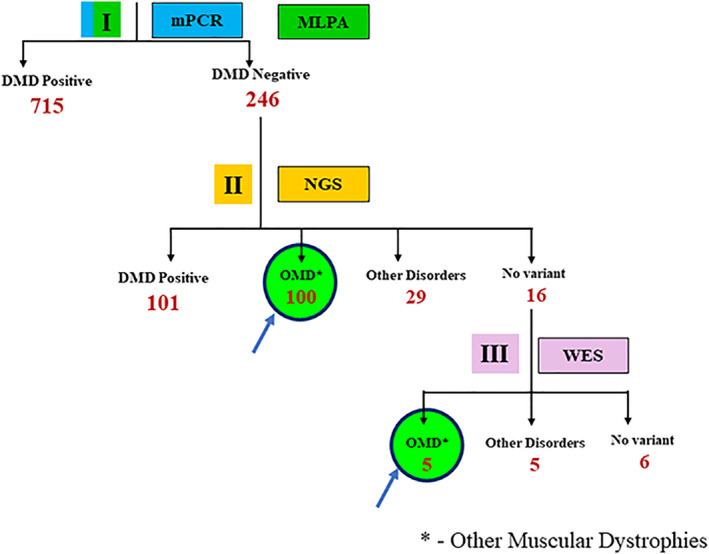
图 1.对 961 名疑似 DMD 患者使用的分子诊断策略发现 105 名患者患有其他肌营养不良症 (OMD)。用于诊断 961 名临床疑似 DMD 患者的算法。mPCR:多重 PCR,MLPA:多重连接依赖性探针扩增;NGS:下一代测序;WES:全外显子组测序。每个步骤的患者数量以红色表示。
全外显子组测序
DNA分离、外显子组文库制备和测序
使用肌营养不良基因检测描述的手动盐析法从全血中分离基因组 DNA 。为了制备文库,将 200 ng Qubit 定量 DNA 片段化为 ~350 bp 插入片段。使用 ~50 Mb Agilent Sure Select 全外显子组面板 (Agilent, Santa Clara, CA, USA) 对文库进行杂交和富集。然后对片段进行末端修复、3′腺苷酸化并与索引接头连接。然后使用接头特异性引物扩增接头连接的片段,然后进行大小选择和纯化以生成 gDNA 文库。使用 Tape Station评估文库的片段大小分布,使用 Qubit(美国赛默飞世尔科技)进行量化,并根据制造商的协议在 Illumina Hiseq 4500(美国加州 Illumina)机器上测序为 2 × 150 bp 双端读数。文库测序的平均测序深度为 ≥80–100×。
数据处理、变异调用和注释
使用 fastq‐mcf(版本 1.04.676)进行质量检查和接头修剪后,将获得的测序读数与人类参考基因组(GRCh37/hg19)比对。对齐的读数进行排序,删除重复读数,并使用 Sentieon(v201808.07)的 GATK 最佳实践流程调用变异。使用 VEP 程序针对基因组的 Ensembl 版本 91 人类基因模型对变异进行基因注释。对变异的等位基因频率进行了注释[群体数据库 GnomAD(v3.0)、1000 基因组、MedGenome 群体特定数据库、计算机预测工具[CADD、PolyPhen‐2、SIFT、Mutation Taster2 和 LRT] 和疾病数据库 [OMIM、ClinVar 和 HGMD]。对具有临床意义的变异进行优先排序,并使用 Varminer(MedGenome 专有变异解释工具)进行分析。除了单核苷酸变异 (SNV) 和小 Indel,还使用 ExomeDepth (v1.1.10) 方法从目标序列数据中检测拷贝数变异 (CNV)。根据测试数据读取深度与匹配的聚合参考数据集的比较,该算法检测 CNV(≥400 bp 缺失和重复)。与疾病表型和遗传相关的基因变异被优先考虑。根据 ACMG 指南对变异进行临床解释。
统计分析
所有统计分析均使用 Microsoft Excel 进行。p值< 0.05 被认为具有显著性。
结果
对 961 名疑似 DMD 患者采用分子诊断策略发现 145 名患者患有其他肌营养不良症 (OMD) 和其他非肌营养不良症
961 名男性患者的初步临床诊断主要在社区初级卫生保健层面进行,主要依据症状,特别强调发病年龄、频繁跌倒、蹒跚步态、小腿肌肉肥大、爬楼梯困难、Gower 征和丧失行走能力的年龄。年龄在 2-35 岁之间且有一种或多种这些临床症状的男性被确定为临床疑似 DMD 患者。正如肌营养不良基因检测之前所报告的,961 名临床疑似 DMD 患者的 DNA 样本接受了方法学算法,采用 mPCR、MLPA 和 NGS 的顺序方案,旨在识别DMD基因中的变异。如图 1所示,在 961 名疑似 DMD 患者中,715 名(74.4%)在使用多重 PCR 和 MLPA 进行 DNA 分析后被 证实患有DMD基因变异,另有 246 名患者在这一轮分析后未检测到变异。这组 246 名疑似 DMD 患者接下来接受了下一代测序 (NGS) 分析,结果发现另外 101 名患者的DMD基因存在小的缺失/重复或点突变。这些相对较小的突变(包括点突变和小的缺失或插入)未被 mPCR 和 MLPA 方法检测到。因此,NGS 分析提高了诊断率,从而将 DMD/BMD 的诊断从 74.4%(mPCR 和 MLPA)确诊到肌营养不良基因检测队列中的总共 84.9% 的患者(图 1)。
15% (145/961) 的患者尽管临床上被怀疑患有 DMD/BMD,但却 没有被发现DMD基因变异。NGS 数据分析进一步显示,145 名患者中有 100 名(图1)患有其他肌营养不良症(OMD;注:为了方便描述,肌营养不良基因检测将其他肌营养不良症 (OMD)定义为没有DMD基因突变,但患有真正的肌营养不良症,并且具有已知肌营养不良相关基因的变异的患者),包括肢带型肌营养不良症 (LGMD)、Bethlem 肌病和 Emery Dreifuss 肌病等。145 名患者中有 29 名被发现患有其他疾病(非肌营养不良症),并且有一些与 DMD 重叠的临床症状。其中包括诊断为轴突夏科-马里图斯综合征和杆状体肌病等的患者。
NGS 分析进一步显示,肌营养不良基因检测队列中的 145 名患者中有 16 名在肌营养不良基因检测的肌营养不良症 NGS 基因组中检测的基因中没有变异。为了进一步研究这组患者的潜在遗传原因,肌营养不良基因检测对他们的 DNA 进行了全外显子组序列 (WES) 分析。肌营养不良基因检测的分析显示,另外五名患者患有其他肌营养不良症 (OMD),而最初的 NGS 分析未能发现这些疾病,这可能是由于两种方法之间的程序差异。发现两名其他肌营养不良症 (OMD)患者有两个不同基因的突变(患者 1;SGCB和UBA1;患者 2:SYNE2和ANO5)(表 1;绿色阴影)。另外 16 名患者中的 5 名被诊断出患有其他疾病,包括先天性肌强直、夏科-马里-图斯病和布罗迪肌病等(表 1)。目前有六名患者仍未得到确诊,且在查询 WES 肌营养不良症基因小组时未发现可识别的变异(表 1)。
表 1.16 个患者样本的全外显子组测序 (WES) 数据
|
序号
|
该基因是否在 NGS 面板中测试过? | 基因 | 疾病类别 | 疾病名称 | 所检测出的突变 | 基因突变的致病性 |
| 1 | 是的 | SGCB | OMD | 肢带型肌营养不良症-2E | c.‐60_(429+1_430–1){0} | 可能致病 |
| 不 | UBA1 | OMD | X连锁脊髓性肌萎缩症2 | c.334C>T | 意义不明 | |
| 2 | 不 | SYNE2 | OMD | Emery‐Dreifuss 肌营养不良症 5 | c.7076G>T | 意义不明 |
| 是的 | ANO5 | OMD | 肢带型肌营养不良症-2 L | c.827A>T | 意义不明 | |
| 3 | 是的 | COL6A3 | OMD | 乌尔里希先天性肌营养不良症-1;贝特莱姆肌病-1 | c.5645C>T | 意义不明 |
| 4 | 是的 | SGCG | OMD | 肢带型肌营养不良症-2C | c.(297+1_298–1)_(385+1_386‐1)del | 可能致病 |
| 5 | 是的 | FKTN | OMD | 伴有脑和眼部异常的先天性 MD‐ 肌营养不良症 / 不伴有智力发育障碍的先天性 MD‐ 肌营养不良症 / LGMD—肌营养不良症 | c.436C>T | 意义不明 |
| 6 | 不 | CLCN1 | 其他的 | 先天性肌强直 | c.2467C>T | 致病 |
| 7 | 不 | MICU1 | 其他的 | 伴有锥体外系征象的肌病 | c.91C>T | 致病 |
| 8 | 不 | SH3TC2 | 其他的 | 夏科-马里-图斯病 4C 型 | c.3327+2T>C | 致病 |
| 9 | 不 | ATP2A1 | 其他的 | 布罗迪性肌病 | 约1808C>T | 意义不明 |
| 不 | NEFH | 其他的 | 轴索性夏科-马里-图斯病 2 型 CC | c.74A>G | 意义不明 | |
| 10 | 不 | CHRND | 其他的 | 快通道先天性肌无力综合征-3B/先天性肌无力综合征-3C | c.259C>T | 意义不明 |
| 11 | 不适用 | 无变体 | 无变体 | 无变体 | 无变体 | 无变体 |
| 12 | 不适用 | 无变体 | 无变体 | 无变体 | 无变体 | 无变体 |
| 13 | 不适用 | 无变体 | 无变体 | 无变体 | 无变体 | 无变体 |
| 14 | 不适用 | 无变体 | 无变体 | 无变体 | 无变体 | 无变体 |
| 15 | 不适用 | 无变体 | 无变体 | 无变体 | 无变体 | 无变体 |
| 16 | 不适用 | 无变体 | 无变体 | 无变体 | 无变体 | 无变体 |
大多数OMD患者患有常染色体隐性遗传病
通过 NGS 和 WES 分析发现,105 名其他肌营养不良症 (OMD)患者中 78 名 (74.2%) 患有常染色体隐性遗传 (AR) 疾病(表 2)。虽然肢带型肌营养不良症 (LGMD) 2E 在该类患者中最为常见,为 37.1%,但 LGMD 2A (14%) 和 LGMD 2B (11.5%) 也很常见(表 2)。发现的其他 LGMD 包括 LGMD 2C、2D、2F 和 2Q。这一发现(表 2)并不令人惊讶,因为近亲结婚和内婚是 AR 疾病传播的基础,在一些地区很常见。在这方面,在肌营养不良基因检测 105 例其他肌营养不良症 (OMD)病例中,肌营养不良基因检测发现 58 例病例为近亲夫妇所生,其中 45 例患有 AR LGMD,其中 LGMD2E 最为常见(21/45)。在家族史方面,105 名患者中有 23 名有家族病史(表 3)。有趣的是,105 名患者中有 18 名既有家族病史,也是近亲所生。其中,LGMD 2E 再次占主导地位,有五个家庭既有家族病史,也有近亲所生。
表 2.已识别的其他肌营养不良症的分类
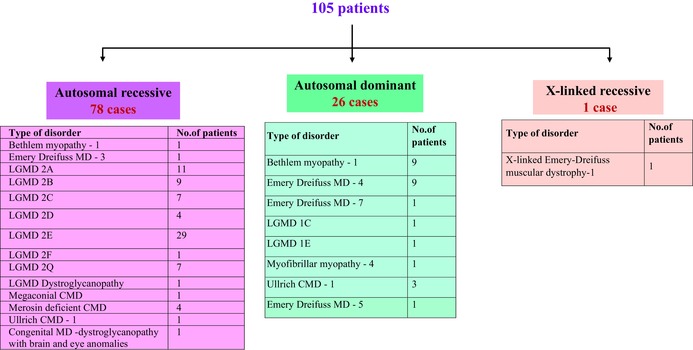 |
表 3.常染色体隐性遗传、常染色体显性遗传和 X 连锁隐性遗传疾病患者的数据汇总
|
序号
|
疾病名称 | 患者人数 | 遗传模式 | 发病年龄 | 门诊病人数量 | 无法走动的病人数量 | 丧失行走能力的年龄 | 有家族史的数字 | 有血缘关系的数字 |
| 1 | Bethlem | 10 | AD – 9 | 1 to 15岁 | 8 | 1 | 11岁 | 2 | 3 |
| AR – 1 | |||||||||
| 2 | Emery | 13 | AD – 11 | 2 to 18岁 | 7 | 4 | 3 to 12岁 | 5 | 7 |
| AR – 1 | |||||||||
| XR – 1 | |||||||||
| 3 | LGMD 2A | 11 | AR – 11 | 6月 to 18岁 | 8 | 2 | 19 to 20岁 | 2 | 5 |
| 4 | LGMD 2B | 9 | AR – 9 | 3 to 32岁 | 8 | 1 | 30岁 | 2 | 7 |
| 5 | LGMD 2E | 29 | AR – 29 | 3 to 18 | 19 | 8 | 8 to 19岁 | 6 | 21 |
| 6 | LGMD 2Q | 7 | AR – 7 | 2 to 6 | 4 | 1 | 14岁 | 2 | 2 |
| 7 | 其他轻微肌营养不良症 | 26 | AR – 20 | 6月 to 10岁 | 22 | 1 | 11岁 | 4 | 13 |
| AD – 6 | |||||||||
| 总数 | 105 | 23 | 58 | ||||||
缩写:AD,常染色体显性遗传;AR,常染色体隐性遗传;XR,X连锁隐性遗传。
105 名患者中,有 26 例为常染色体显性 (AD) 遗传模式的其他肌营养不良症 (OMD)患者。其中,Bethlem 肌病和 Emery Dreifuss 肌营养不良症最为常见(各占 34.6%)(表 2)。仅发现一名患有 X 连锁 AR 疾病的患者(表 2;X 连锁 Emery-Dreifuss 肌营养不良症-1)。
大多数其他肌营养不良症 (OMD)患者中发现的新变异
在 105 名患有所列其他肌营养不良症 (OMD)的患者中,共发现 78 种基因变异(表 4)。有趣的是,在 78 种基因变异中,有 44 种(56.4%)被发现是新变异。所有新变异都已存入 Global Variome 共享 LoVD 数据库中,并标记为“新变异(2021 年)”
表 4.105 个其他肌营养不良症 (OMD)突变状态汇总显示共有 44 个新变异
|
疾病
|
基因 | 新型变体 | 此前报道 | 总变体数 |
| LGMD 2A | CAPN3 | 3 | 5 | 8 |
| LGMD 2B | DYSF | 6 | 2 | 8 |
| LGMD 2C | SGCG | 4 | 2 | 6 |
| LGMD 2D | SGCA | 1 | 3 | 4 |
| LGMD 2E | SGCB | 4 | 5 | 9 |
| LGMD 2F | SGCD | 1 | 0 | 1 |
| LGMD 2Q | PLEC | 6 | 0 | 6 |
| LGMD 1C | CAV3 | 0 | 1 | 1 |
| LGMD 1 E | DNAJB6 | 1 | 0 | 1 |
| LGMD 肌营养不良症 | POMT2 | 0 | 1 | 1 |
| 贝特莱姆肌病 | COL6A1, COL6A2, COL6A3 | 4 | 6 | 10 |
| Emery‐Dreifuss 肌营养不良症 3 | LMNA | 0 | 1 | 1 |
| Emery‐Dreifuss 肌营养不良症 4 | SYNE1 | 5 | 4 | 9 |
| Emery‐Dreifuss 肌营养不良症 5 | SYNE2 | 1 | 0 | 1 |
| Emery‐Dreifuss 肌营养不良症 7 | TMEM43 | 0 | 1 | 1 |
| 伴有脑和眼部异常的先天性肌营养不良症‐肌营养不良蛋白病 | FKTN | 0 | 1 | 1 |
| 巨尖肌型先天性肌营养不良症 | CHKB | 1 | 0 | 1 |
| 美洛星缺乏型先天性肌营养不良症 1A 型 | LAMA2 | 2 | 1 | 3 |
| 肌原纤维肌病‐4 | LDB3 | 1 | 0 | 1 |
| 乌尔里希先天性肌营养不良症 1 | COL6A1, COL6A2 | 3 | 0 | 3 |
| 乌尔里希先天性肌营养不良症-1;贝特莱姆肌病-1 | COL6A3 | 0 | 1 | 1 |
| X 连锁 Emery-Dreifuss 肌营养不良症-1 | EMD | 1 | 0 | 1 |
| 总数 | 44 | 34 | 78 |
在新变异中,LGMD2Q(n = 6)和 LGMD2B(n = 6)患者中发现的数量最多。鉴于所有变异中有 56.4% 被发现是新变异,因此大多数(46%)被预测为意义不明确的变异 (VUS),27% 的变异被发现致病,其余 27% 的变异可能致病,这并不奇怪。78 种基因变异中有 34 种之前已在文献中报道过(表 4)。
三分之一的其他肌营养不良症 (OMD)患者(32.3%)被诊断患有其他疾病
NGS 和 WES 分析共同显示,105 名患者中有 34 名并未患有真正的肌营养不良症,因此被归类为“其他疾病”患者,如表 5所示。该组患者中的大多数(19/34)患有常染色体隐性遗传病,其中最常见的是先天性肌无力综合征(7/19),这是一组以肌肉无力为特征的遗传性疾病,且在体力消耗后会加剧。杆状体肌病是第二常见的疾病(5/19)。杆状体肌病是一组异质性的先天性肌病,由至少 12 个基因的从头突变引起,其中最常见的是α‐肌动蛋白(ACTA1)和肌动蛋白(NEB )。症状范围广泛,以肌肉无力和肌张力低下为主要症状。19 名患者中有 3 名患有常染色体隐性遗传的夏科-马里-图斯 (CMT) 病,而 14 名患者中有 2 名患有常染色体显性遗传的 CMT (表 5 )。CMT 代表一组异质性神经病变,其主要特征是肌肉无力和肌肉尺寸减小。
表 5.其他疾病患者的分类
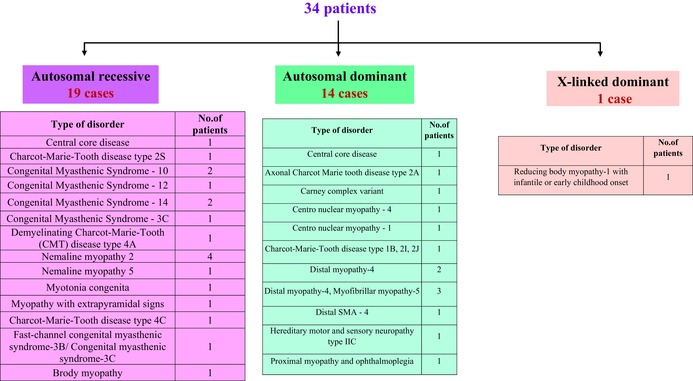 |
在被确诊患有常染色体显性遗传病的 14 名患者中,在其他疾病类别中,有 5 名患者患有远端肌病,这是一组原发性肌肉疾病,与其他肌营养不良症一样,表现出肌肉无力和肌纤维逐渐丧失。还发现了一例由佳学基因检测进行检测与分析 X 连锁显性减退体肌病-1 病例,发病时间为婴儿期或幼儿期 (表 5 )。这类“其他疾病”的几种临床症状与 DMD 的临床症状重叠,将在其他地方详细阐述(手稿正在准备中)。
其他肌营养不良症(OMD)患者的分析:常染色体隐性肢带型肌营养不良症(LGMD)
肢带型肌营养不良症 2A 型 (LGMD2A/LGMDR1) (MIM# 253600)
LGMD2A 最近被归类为 LGMDR1,是一种进行性肌病,表现为缺乏一种骨骼肌特异性的钙依赖性钙蛋白酶半胱氨酸蛋白酶家族异构体,即钙蛋白酶 3 ( CAPN3 )在肌营养不良基因检测研究的 78 名常染色体隐性其他肌营养不良症 (OMD)男性患者中,有 11 名被发现存在与 LGMD2A 相关的CAPN3基因变异。在这 11 名患者中,总共发现了 8 种CAPN3基因变异,其中每名患者都有 2 名发现了 3 个点突变 (c.2003T>G、c.946-2A>G 和 c.1343G>A)(表 6;参见彩色单元格)。此外,发现 3/8 的突变体是新的(表 6)。在肌营养不良基因检测这个队列中,发病平均年龄为 11.2±5.6 岁,诊断年龄为 18.8±5.9 岁,范围为 8-27 岁。在这个队列中,发病和诊断之间的间隔为 7.1 年。
表 6.11 名患有 LGMD2A(LGMDR1)且CAPN3基因发生突变的患者
|
序号
|
发病年龄 | 诊断年龄 | 血缘 | 是否有家族史 | 已识别变体 | 变体分类 | 接合性 | 已报道/新发现 |
| 1 | 18 | 23 | 是 | 是 | c.608C>T | 可能致病 | 纯合 | 已报道 |
| 2 | 12 | 24 | 是 | 否 | c.1273A>T | 可能致病 | 纯合 | 新发现 |
| 3 | 13 | 15 | 否 | 否 | c.1333G>C | 致病 | 纯合 | 已报道 |
| 4 | 无数据 | 11 | 否 | 否 | c.2050+1G>A | 致病 | 纯合 | 已报道 |
| 5 | 3 | 17 | 否 | 否 | c.2033A>T | 意义不明 | 杂合子 | 新发现 |
| 6 | 9 | 27 | 否 | 否 | c.2003T>G | 意义不明 | 纯合 | 新发现 |
| 7 | 6个月 | 8 | 是 | 否 | c.2003T>G | 意义不明 | 纯合 | |
| 8 | 13 | 15 | 否 | 否 | c.946‐2A>G | 致病 | 纯合 | 已报道 |
| 9 | 15 | 16 | 否 | 否 | c.946‐2A>G | 致病 | 纯合 | |
| 10 | 13 | 24 | 是 | 是 | c.1343G>A | 致病 | 纯合 | 已报道 |
| 11 | 15 | 20 | 是 | 否 | c.1343G>A | 致病 | 纯合 |
注意:彩色细胞表示相同的CAPN3基因变体。45% 血缘关系和 18% 家族史。
肢带型肌营养不良症 2B/R2(MIM# 253601)
LGMD2B/R2 属于一类被称为“dysferlinopathies”的疾病,该疾病是由DYSF基因突变导致 dysferlin 蛋白水平不足引起的。LGMD2B 被认为是 LGMD 的较轻形式,因为 LGMD2B 的发病年龄虽然各不相同,但通常相对较晚,在 20-30 岁之间发病。LGMD2B 的特征是骨盆和肩带肌肉无力和萎缩。在所有 LGMD 中,LGMD2B 的进展最慢。
9 名疑似 DMD 患者经 NGS 分析确诊为 LGMD2B/R2,其中 6 名患者DYSF基因发生新突变(表 7)。一对无亲缘关系的患者具有相同的DYSF基因突变(c.1256G>C;表 7)。平均发病年龄为 17.8±8.8 岁,而该组患者的诊断年龄为 31.5±10.7 岁(范围为 19-56 岁)。发病与诊断之间相差 13.8 年。
表 7.9 名 LGMD2B(LGMDR2)患者发生DYSF基因突变
| 序号 | 发病年龄 | 诊断年龄 | 血缘 | 家史 | 已识别变体 | 变体分类 | 接合性 | 已报道/新发现 |
|---|---|---|---|---|---|---|---|---|
|
1 |
22 |
33 |
不 |
不 |
c.2625G>A |
致病 |
纯合子 |
新发现 |
|
2 |
19 |
27 |
不 |
不 |
c.6131G>T |
可能致病 |
纯合子 |
新发现 |
|
3 |
32 |
35 |
是的 |
不 |
c.4110_4112del |
致病 |
纯合子 |
新发现 |
|
4 |
25 |
56 |
是的 |
是的 |
c.3321_3329delCCGCCCGCCG |
意义不明 |
纯合子 |
新发现 |
|
5 |
3 |
21 |
是的 |
不 |
c.2266C>T |
可能致病 |
纯合子 |
新发现 |
|
6 |
10 |
19 |
是的 |
不 |
c.5743G>A |
意义不明 |
纯合子 |
已报道 |
|
7 |
10 |
30 |
是的 |
不 |
约395C>A |
意义不明 |
杂合子 |
新发现 |
|
8 |
19 |
35 |
是的 |
是的 |
c.1256G>C |
可能致病 |
纯合子 |
已报道 |
| 9 | 20 | 28 | 是的 | 不 | c.1256G>C | 可能致病 | 纯合子 |
注:彩色单元格表示相同的DYSF基因变体。78% 血缘关系和 22% 家族史。
肢带型肌营养不良症 LGMD2Q/R17 (MIM# 613723)
七名患者经 NGS 证实具有 Plectin(PLEC)基因变异,从而可能诊断为 LGMD2Q。在这组患者中观察到了 与PLEC基因相关的多种突变类型。虽然发现患者 1 和 6(表8)携带PLEC基因杂合突变,但发现患者 3、4、5 和 7 具有PLEC基因复合杂合变异(表 8)。此外,患者 2、3 和 5除了PLEC基因杂合变异外,还具有LAMA2和DYSF基因变异(表 8)。该组患者的发病年龄为 4.3+/- 1.5 岁,而诊断年龄为 14+/-7 岁。在肌营养不良基因检测的队列中, PLEC基因中发现的 7 种变异中有 6 种是新的(表 8),其中 7 名患者中有 2 名具有相同的PLEC变异(c.6359G>A),所有PLEC基因变异均归类为 VUS。
表 8.7 名 LGMD2Q(LGMDR17)患者存在PLEC基因突变
| 序号 | 发病年龄 | 诊断年龄 | 血缘 | 家史 | 杂合性 | 突变 1 | 突变 2 | 突变 3 |
|---|---|---|---|---|---|---|---|---|
| 1 | 6 | 15 | 不 | 是的 | 杂合子 | PLEC ;外显子 11;c.1570G>A | 没有任何 | 无 |
| 2 | 6 | 无数据 | 不 | 不 | 杂合子 | PLEC ;外显子 14;c.2078G>A | LAMA2 ;外显子 14;c.1963C>T | 无 |
| 3 | 3 | 15 | 是的 | 不 | 杂合子 | PLEC ;外显子 32;c.10589C>T | PLEC ;外显子 31;c.5008C>T | DYSF ;外显子 34;c.3779G>A |
| 4 | 2 | 24 | 不 | 不 | 杂合子 | PLEC ;外显子 32;c.8922G>C | PLEC ;外显子 28;c.4198G>A | 无 |
| 5 | 无数据 | 5 | 是的 | 不 | 杂合子 | PLEC ;外显子 32;c.11026C>T | PLEC ;外显子 15;c.2170C>T | DYSF ;内含子 34;c.3898-4C>G |
| 6 | 5 | 18 | 不 | 是的 | 杂合子 | PLEC ;外显子 31;c.6359G>A | 没有任何 | 无 |
| 7 | 4 | 7 | 不 | 不 | 杂合子 | PLEC ;外显子 31;c.6359G>A | PLEC ;外显子 31;c.6284C>T | PLEC ;外显子 23;c.3206A>G |
注:彩色单元格表示相同的PLEC基因变体。28%血缘关系和28%家族史。
3.5.4. 肢带型肌营养不良症 2E/ (LGMD2E/R4, MIM# 604286)
在 105 名因就诊症状而被误诊为 DMD 的患者中,29/105 (27.6%) 名患者通过 NGS 分析被准确诊断为患有 LGMD2E(表 9 )。LGMD2E 是一种常染色体隐性遗传病,由 β‐肌聚糖 ( SGCB )基因突变引起。
表 9.LGMD2E:29 名 LGMD2E(LGMDR4)患者,SGCB 发生突变,并具有临床特征
| 序号 | 已识别的基因序列变化 | 分类—NGS | 诊断年龄 | 是否有血缘关系 | 是否有家家史 | 发病年龄 | 行走状况 | 丧失劳动能力年龄 | 接合性 | 报道/新发现 |
| 1 | c.655A>T | 致病 | 10 | 是的 | 不 | 3 | 可行走 | 不适用 | 纯合子 | 新发现 |
| 2 | c.621+1G>T | 致病 | 14 | 无法使用 | 无法使用 | 10 | 不能行走 | 13 | 纯合子 | 已报道 |
| 3 | c.499G>A | 可能致病 | 6 | 不 | 不 | 6 | 可行走 | 不适用 | 纯合子 | 已报道 |
| 4 | 约244-1G>A | 可能致病 | 8 | 不 | 不 | 7 | 可行走 | 不适用 | 纯合子 | 新发现 |
| 5 | 约 490A>G | 可能致病 | 32 | 是的 | 不 | 18 | 可行走 | 不适用 | 纯合子 | 新发现 |
| 6 | c.355A>T | 可能致病 | 22 | 不 | 不 | 10 | 不能行走 | 19 | 纯合子 | 已报道 |
| 7 | c.‐60_(429+1_430–1){0} | 可能致病 | 11 | 是的 | 是的 | 5 | 不能行走 | 10 | 纯合子 | 新发现 |
| 8 | c.572delT | 致病 | 8 | 是的 | 不 | 6 | 可行走 | 不适用 | 纯合子 | 已报道 |
| 9 | c.572delT | 致病 | 19 | 不 | 不 | 9 | 不能行走 | 14 | 杂合子 | |
| 10 | c.572delT | 致病 | 12 | 不 | 不 | 9 | 可行走 | 不适用 | 杂合子 | |
| 11 | c.572del | 致病 | 8 | 是的 | 不 | 3 | 可行走 | 不适用 | 纯合子 | |
| 12 | c.544A>C | 意义不明 | 9 | 不 | 不 | 7 | 可行走 | 不适用 | 纯合子 | 已报道 |
| 13 | c.544A>C | 意义不明 | 13 | 是的 | 不 | 5 | 不能行走 | 9 | 纯合子 | |
| 14 | c.544A>C | 意义不明 | 10 | 是的 | 无法使用 | 8 | 不能行走 | 9 | 纯合子 | |
| 15 | c.544A>C | 意义不明 | 12 | 是的 | 不 | 7 | 可行走 | 不适用 | 纯合子 | |
| 16 | c.544A>C | 意义不明 | 7 | 是的 | 不 | 6 | 可行走 | 不适用 | 纯合子 | |
| 17 | c.544A>C | 意义不明 | 8 | 是的 | 不 | 6 | 可行走 | 不适用 | 纯合子 | |
| 18 | c.544A>C | 意义不明 | 5 | 是的 | 不 | 3 | 可行走 | 不适用 | 杂合子 | |
| 19 | c.544A>C | 意义不明 | 11 | 是的 | 不 | 7 | 可行走 | 不适用 | 纯合子 | |
| 20 | c.544A>C | 意义不明 | 7 | 是的 | 是的 | 6 | 可行走 | 不适用 | 纯合子 | |
| 21 | c.544A>C | 意义不明 | 7 | 是的 | 不 | 4 | 可行走 | 不适用 | 纯合子 | |
| 22 | c.544A>C | 意义不明 | 11 | 是的 | 是的 | 无法使用 | 无法使用 | 无法使用 | 纯合子 | |
| 23 | c.544A>C | 意义不明 | 11 | 是的 | 不 | 6 | 不能行走 | 10 | 纯合子 | |
| 24 | c.544A>C | 意义不明 | 4 | 是的 | 是的 | 无法使用 | 无法使用 | 无法使用 | 杂合子 | |
| 25 | c.544A>C | 意义不明 | 10 | 不 | 是的 | 7 | 可行走 | 不适用 | 纯合子 | |
| 26 | c.544A>C | 意义不明 | 10 | 是的 | 不 | 5 | 可行走 | 不适用 | 纯合子 | |
| 27 | c.544A>C | 意义不明 | 8 | 是的 | 是的 | 4 | 可行走 | 不适用 | 纯合子 | |
| 28 | c.544A>C | 意义不明 | 8 | 是的 | 不 | 无法使用 | 可行走 | 不适用 | 纯合子 | |
| 29 | c.544A>C | 意义不明 | 11 | 是的 | 不 | 3 | 不能行走 | 8 | 纯合子 |
注:彩色单元格表示相同的SGCB基因变体。72% 血缘关系和 21% 家族史。
虽然在 29/105 名SGCB基因突变患者中共发现 9 种变异(表 9和图 2a中的箭头),但在该 LGMD2E 患者队列中遇到了两组常见变异。c.544A>C 点变异出现在 18/29 (62%) 名无关患者中(图 2a,红色箭头),而 c.572delT 移码变异出现在 4 名患者中(图 2a,粉色箭头)。这两种变异均曾被报道过,但并未出现在来自单个部位的多名患者样本中。虽然前一种变异被预测可能致病,但后者已被发现具有致病性(表 9)。
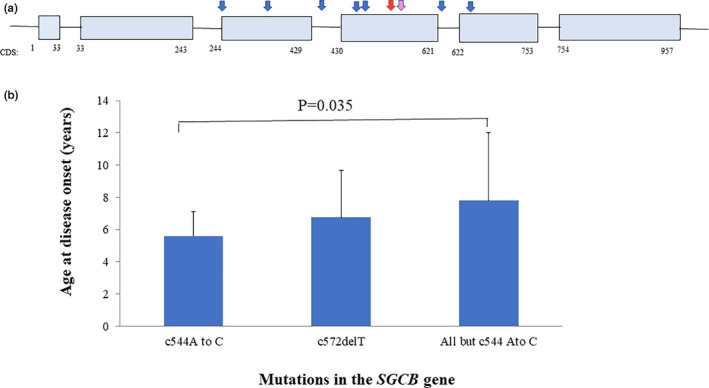
图 2.LGMD2E 患者SGCB基因的突变和功能特征。(a)SGCB 基因的组织显示六个外显子,cDNA 坐标编号如下。蓝色箭头表示患者中发现的单个突变的位置。红色箭头表示最常见的 c.544A>C 变体;粉色箭头:显示 c572delT 变体的位置。外显子 1 和 2 编码 IC 结构域,外显子 2 和 3 编码跨膜结构域;外显子 3、4、5 和 6 编码 EC 结构域,外显子 1 和 6 包含 5′ 和 3′ 非编码序列。(b)根据 LGMD2E 患者发病年龄比较 SGCB 基因中不同突变的疾病严重程度。除 544 a 到 C 外,其他均指除 c.544A>C 之外的所有变体。计算出的p值为 0.035 被认为具有统计学意义。
肌营养不良基因检测发现 29 名患者发病年龄为 6.5+/-3.1 岁。失去行走能力的年龄为 11.5+/-3.7 岁,似乎低于之前报告的年龄)。为了确定是否可以从这组患者中收集到基因型与表型的相关性,肌营养不良基因检测比较了 c.544A>C 变异患者(n = 18)与 c572delT(n = 4)和其他统称的变异患者(n = 7)的发病年龄。如图 2b所示,c.544A>C 变异患者的发病年龄为 5.6+/-1.5 岁,而 c.572delT 变异患者的发病年龄为 6.8+/-2.9 岁。虽然这两种变异的患者在这项指标上没有统计学上的显著差异,但变异 c.544A>C 与队列中其他变异之间存在显著差异(图 2b:p = 0.035)。鉴于 c.544A>C 和 c.572delT 变异之间的发病年龄没有统计学差异,并且后者已被预测为致病,肌营养不良基因检测的数据支持将前者从意义不明的变异重新归类为致病变异。
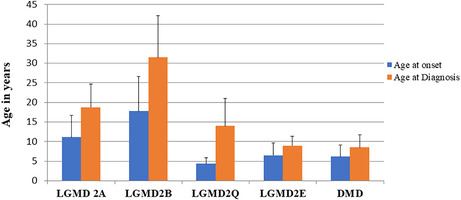
鉴于肌营养不良基因检测临床大数据分析中发现的所有 LGMD 均被误诊为 DMD/BMD,肌营养不良基因检测试图确定是否可以根据发病年龄将 LGMD 与 DMD 区分开来。如图 3所示,肌营养不良基因检测之前对 961 名患者的研究发现,DMD 患者的平均发病年龄为 6.17+/− 3 岁。相比之下,LGMD2A 的发病年龄为 11.2+/−5.6(比 DMD 高 1.8 倍),LGMD2B 为 17.8+/−8.8(比 DMD 高 2.9 倍),LGMD2Q 为 4.3+/−1.5(比 DMD 低 0.7 倍或 30%),LGMD2E 为 6.5+/−3.1(与 DMD 相比无变化)。基于这一单一标准,很明显,在肌营养不良基因检测的队列中,LGMD2E 的严重程度与 DMD 最接近,其次是 LGMD 2Q。
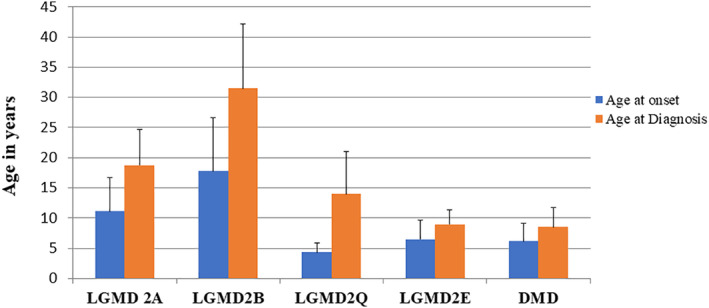
图 3. LGMD 的发病年龄和诊断年龄与 DMD 的比较。诊断为 LGMD、2A、2B、2E、2Q 和 DMD 的患者的发病年龄和诊断年龄(以岁为单位)的比较。
比较该队列中 LGMD 患者的发病年龄和诊断年龄(图 3),并与 DMD 进行比较,结果表明,LGMD2B 患者的两项指标之间的差异最大(13.8 岁),其次是 LGMD2Q(9.7 岁)和 LGMD2A(7.1 岁)。临床上最严重的肌营养不良症 DMD(3.9 岁)和 LGMD2E(4.2 岁)的发病年龄和诊断年龄之间的差异似乎最小。
常染色体显性肌病
Bethlem 肌病 (MIM# 158810) 和 Emery Driefuss 肌营养不良症 (MIM# 616516)
在 105 名疑似 DMD 患者的 NGS 筛查中,发现了 10 例 Bethlem 肌病和 4 例 Ullrich 先天性肌营养不良症 (UCMD;MIM# 254090)(表 2)。与表现为严重表型的 Ullrich 先天性肌营养不良症相比,Bethlem 肌病是一种较轻的先天性常染色体显性肌病。这两种疾病都是由编码 VI 型胶原蛋白的基因突变引起的,包括COL6A1(a1 链)、COL6A2(a2 链)和COL6A3(a3 链)。胶原蛋白 VI 是细胞外基质的重要组成部分,可生成与细胞和周围基底膜相关的微纤维网络。已知 IV 型胶原蛋白的突变会影响结缔组织和肌肉,患者常常出现 Gower 征、足尖行走和关节挛缩。
10 名确诊患有 Bethlem 肌病的患者中,有 4 名患有COL6A1基因突变,5 名患者患有COL6A3基因突变,1 名患者患有COL6A2基因突变(表 10)。10 个已识别的变异中有 4 个是新发现的。这一小群患者的发病年龄和诊断年龄各不相同,平均年龄为 10.6+/-7.86 岁(范围:2-23 岁,表 10)。
表 10.
10 名患有 Bethlem 肌病 (LGMDD5) 且COL6A基因发生突变的患者
| 序号 | 发病年龄 | 诊断年龄 | 血缘 | 家史 | 基因 | 已识别变体 | 分类—NGS | 接合性 | 报道/新发现 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 没有数据 | 2 | 不 | 不 | COL6A1 | c.1022G>A | 可能致病 | 杂合子 | 已报道 |
| 2 | 1 | 11 | 不 | 不 | COL6A1 | c.2348G>A | 意义不明 | 杂合子 | 已报道 |
| 3 | 没有数据 | 3 | 不 | 不 | COL6A1 | 约 979A>G | 意义不明 | 杂合子 | 新发现 |
| 4 | 2 | 8 | 不 | 不 | COL6A1 | c.928_930del | 可能致病 | 杂合子 | 已报道 |
| 5 | 5 | 23 | 不 | 不 | COL6A2 | c.2611G>A | 可能致病 | 纯合子 | 已报道 |
| 6 | 15 | 23 | 不 | 不 | COL6A3 | 约2030G>A | 可能致病 | 杂合子 | 已报道 |
| 7 | 3 | 4 | 是的 | 不 | COL6A3 | c.5825C>T | 意义不明 | 杂合子 | 已报道 |
| 8 | 10 | 14 | 是的 | 是的 | COL6A3 | c.7600T>G | 可能致病 | 杂合子 | 新发现 |
| 9 | 4 | 14 | 是的 | 是的 | COL6A3 | c.4405G>C | 意义不明 | 杂合子 | 新发现 |
| 10 | 3 | 4 | 不 | 不 | COL6A3 | c.5917+2T>C | 可能致病 | 杂合子 | 新发现 |
注:30%有血缘关系,20%有家族史。
金刚砂肌营养不良症(MIM# 616516、MIM# 612998、MIM# 612999、MIM# 614302)
根据 Lamin A/C ( LMNA ; n = 1)、Nesprin 1 ( SYNE1 ; n = 9)、Nesprin 2 ( SYNE2 ; n = 1) 和 LUMA ( TMEM43 ; n = 1) 基因中发现的变异,12 名患者被诊断患有 Emery‐Driefuss 肌营养不良症 (EDMD)(表 11)。EDMD 是一种 X 连锁常染色体显性肌营养不良症,常见症状为肌肉萎缩、关节挛缩、心脏传导异常和心肌病。EDMD 属于一类称为核膜病或核纤层蛋白病的疾病,包括一组不断增长的与核膜基因突变有关的人类遗传性疾病。根据相关的突变基因,OMIM 目前可识别出七种亚型,包括 EDMD1 至 7。然而,超过 60% 的 EDMD 患者没有可识别的EMD(emerin)或LMN突变,这是两种最常见的 EMDA 相关突变基因。肌营养不良基因检测队列中的患者被分为 EDMD3 ( n = 1)、EDMD4 ( n = 9)、EDMD5 ( n = 1) 和 EDMD 7 ( n = 1)(表 11 )。有趣的是,在肌营养不良基因检测的 EDMD 队列中发现的 50% 的变异是新发现的。肌营养不良基因检测估计SYNE1基因变异的 EDMD4 患者的诊断年龄为 17.7 +/−12.6 岁(表 11,范围为 8-37 岁)。
表 11.12 名 Emery‐Dreifuss 肌营养不良症患者
| 序号 | 发病年龄 | 诊断年龄 | 是否有血缘关系 | 是否有家族史 | 疾病 | 基因 | 已识别变体 | 分类—NGS | 接合性 | 报道/新发现 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 无法使用 | 无法使用 | 不 | 不 | EDMD‐3 | LMNA | c.674G>A | 致病 | 纯合子 | 已报道 |
| 2 | 无法使用 | 14 | 是的 | 不 | EDMD‐4 | SYNE1 | c.16671T>A | 意义不明 | 杂合子 | 新发现 |
| 3 | 3 | 二十八 | 是的 | 是的 | EDMD‐4 | SYNE1 | c.3752G>A | 意义不明 | 杂合子 | 新发现 |
| 4 | 8 | 8 | 是的 | 是的 | EDMD‐4 | SYNE1 | c.13089A>T | 意义不明 | 杂合子 | 新发现 |
| 5 | 5 | 8 | 不 | 不 | EDMD‐4 | SYNE1 | c.3962T>C | 意义不明 | 杂合子 | 新发现 |
| 6 | 5 | 11 | 不 | 不 | EDMD‐4 | SYNE1 | c.3656C>T | 意义不明 | 杂合子 | 已报道 |
| 7 | 2 | 8 | 无法使用 | 无法使用 | EDMD‐4 | SYNE1 | c.21222A>C | 意义不明 | 杂合子 | 新发现 |
| 8 | 2 | 9 | 不 | 不 | EDMD‐4 | SYNE1 | c.7611G>T | 意义不明 | 杂合子 | 已报道 |
| 9 | 2 | 37 | 是的 | 是的 | EDMD‐4 | SYNE1 | 约19692+3G>A | 意义不明 | 杂合子 | 已报道 |
| 10 | 18 | 37 | 不 | 不 | EDMD‐4 | SYNE1 | c.25907A>G | 意义不明 | 杂合子 | 已报道 |
| 11 | 7 | 16 | 是的 | 是的 | EDMD‐5 | SYNE2 | c.7076G>T | 意义不明 | 杂合子 | 新发现 |
| 12 | 3 | 15 | 是的 | 不 | EDMD‐7 | TMEM43 | c.488G>A | 意义不明 | 杂合子 | 已报道 |
注:50% 是血缘关系,33% 是家族史。
NGS 分析后发现患者中存在多种同时发生的其他肌营养不良症 (OMD)基因变异
如图 4a和 LOVD 数据库中的数据所示,肌营养不良基因检测 105 名其他肌营养不良症 (OMD)患者中的大多数 (56%) 都携带一个真正的其他肌营养不良症 (OMD)基因变异。有趣的是,大约三分之一 (27.6%) 的患者携带两个变异,而 14.3% 的患者携带三个变异。两名患者 (1.9%) 被发现各有四个变异。其中第一例患者在LAMA2基因中存在两种变异(外显子 15;c.2131_2134dup 4 bp 插入和外显子 60;c.8396C>G),两种变异均被归类为可能致病,并且在PLEC基因中存在两种错义变异(外显子 31;c.4757G>A 和外显子 32;c.13172C>T;均被归类为 VUS)。第二例患者在SYNE1基因中存在一种错义变异(外显子 87;c.16671T>A;VUS),在SGCA基因中存在一种错义变异(外显子 9;c.1109G>A;VUS),并且在NEB基因中存在两种错义变异(外显子 134;c.20419T>C 和外显子 168;c.23969C>T,均为 VUS)。诊断时,第一名患者 14 岁,可以行走;第二名患者 13 岁。撰写本报告时,两名患者分别为 27 岁和 29 岁,但目前行走状况未知。
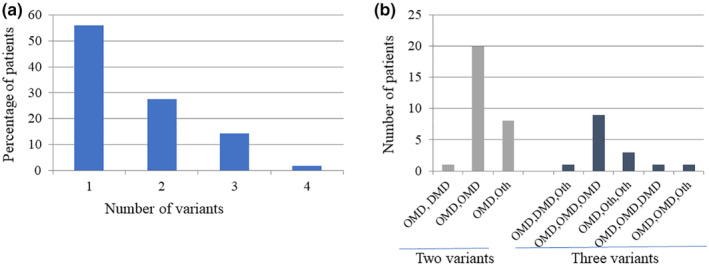
图 4.带有与 OMD、DMD 或其他疾病相关的基因的两个或多个变异的患者。 (a) 队列中带有一个、两个、三个或四个变异的患者百分比。 (b) 带有两个或三个变异且在真正的基因中带有特定变异的患者数量,这些变异是 DMD、OMD(其他肌营养不良症)和/或其他疾病(Oth)。
在具有两种和三种基因变异的患者中(图 4b),大多数具有其他肌营养不良症 (OMD)基因变异(2 种其他肌营养不良症 (OMD)基因变异:20 名患者和 3 种其他肌营养不良症 (OMD)基因变异:9 名患者)。只有少数具有双重变异的患者的临床数据可用(表 12)。两例均为近亲结婚的产物,一例(患者 1)的TMEM43和SYNE1基因有未知意义的变异,另一例(患者 4;表 12 )的SYNE2和ANO5基因有未知意义的变异,这两例患者都不能行走并患有脊柱侧弯。此外,患者 2 是半行走能力,表现为肩胛骨翼状,具有PLEC和LAMA2基因的双重变异。另一方面,患者 3 被发现具有SYNE1和FLNC基因的未知意义的变异。但他仍然没有症状并且看似正常。他曾接受过肌营养不良症基因变异检测,因为他已故的两位叔叔有疑似 DMD 的症状。不幸的是,叔叔们没有接受基因检测。尽管如此,考虑到他的家族病史和其他肌营养不良症 (OMD)相关基因变异的存在,这个人必须保持谨慎。
表 12.患有两种其他肌营养不良症 (OMD)变异且病情严重程度
| ID | 疾病类别 | 疾病类型 | 基因 | 地点 | 已识别变体 | 分类 | 出现症状的年龄 | 诊断时的行走状态 | 丧失行走能力的年龄 | 诊断年龄 | 現在年齡 | 家史 | 血缘 | 具体症状(如果有) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | OMD | Emery‐Dreifuss 肌营养不良症 7 | TMEM43 | 外显子 6 | c.488G>A | 意义不明 | 6 | 不能行走 | 11 | 15 | 二十五 | 不 | 是的 | 脊柱侧弯;患者太瘦 |
| 1b | OMD | Emery‐Dreifuss 肌营养不良症 4 | SYNE1 | 外显子 117 | c.21436C>T | 意义不明 | ||||||||
| 2a | OMD | 肢带型肌营养不良症 2Q 型 | PLEC | 外显子 14 | c.2078G>A | 意义不明 | 9 | 半行走式 | 不适用 | 12 | 21 | 不 | 不 | 第一个症状——肩胛骨翼状 |
| 2b | OMD | 美洛星缺乏型先天性肌营养不良症 1A 型 | LAMA2 | 外显子 14 | 约1963年C>T | 意义不明 | ||||||||
| 3a | OMD | Emery‐Dreifuss 肌营养不良症 4 | SYNE1 | 外显子 78 | c.13089A>T | 意义不明 | 无症状 | 可行走 | 不适用 | 8 | 18 | 是(2 个叔叔) | 是的 | 正常男孩;由于有家族病史,他们接受了检查 |
| 3b | OMD | 远端肌病-4,肌原纤维肌病-5 | FLNC | 外显子 32 | c.5363T>G | 意义不明 | ||||||||
| 4a | OMD | Emery‐Dreifuss 肌营养不良症 5 | SYNE2 | 外显子 45 | c.7076G>T | 意义不明 | 9 | 不能行走 | 十三 | 16 | 二十七 | 不 | 是的 | 脊柱侧弯;肌痛 |
| 4b | OMD | 肢带型肌营养不良症-12 | ANO5 | 外显子 9 | c.827A>T | 意义不明 |
注意:患有两种其他肌营养不良症 (OMD)变体且病情严重程度的患者。
需要注意的是:虽然一种或多种变异对所描述的肌营养不良症的贡献可能很大,但目前还无法就这些突变基因对疾病致病性的贡献得出任何结论。
其他肌营养不良症 (OMD)与 DMD 具有重叠的临床症状,在缺乏基因检测选项的诊所中很容易被误诊
肌营养不良基因检测研究中的大多数患者来农村。由于农村诊所目前不具备使用最先进的 NGS 方法诊断肌营养不良症的能力,因此农村诊所的医生在诊断过程中严重依赖临床表现。因此,在 961 名临床疑似 DMD 患者中,近 15% 经 NGS 和 WES 分析证实患有其他肌营养不良症和其他疾病(见表 2和表5),这并不奇怪。为了确定患者被误诊的原因,肌营养不良基因检测选择了九种常见的 DMD 症状,并询问这些症状在肌营养不良基因检测患者队列中其他经基因确诊的肌营养不良症中出现的频率。根据肌营养不良基因检测在图 5中的观察,与经基因确诊的 DMD 患者相比,所有考虑的 DMD 特异性症状都与肌营养不良基因检测队列中遇到的所有其他肌营养不良症 (OMD)相关,尽管频率有所不同。LGMD2B 和 LGMD2E 患者的运动里程碑延迟症状更为明显,与 DMD 患者相比,贝特莱姆肌病患者中足尖行走更为常见(图 5)。然而,这些结论必须谨慎看待,因为在肌营养不良基因检测的研究中,常染色体隐性遗传病患者数量有限。
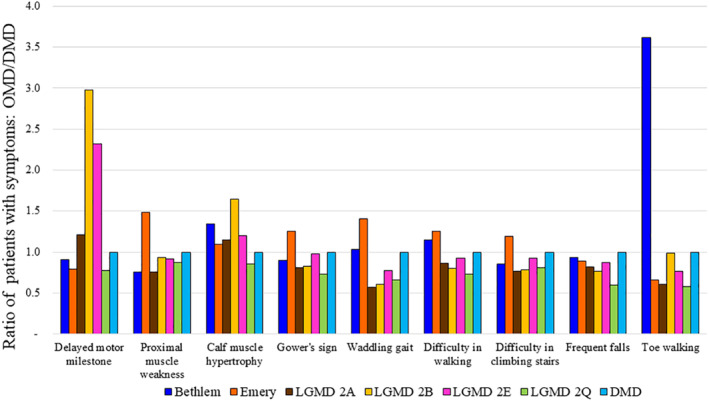
图 5.比较其他肌营养不良症 (OMD)和 DMD 中观察到的症状,发现存在明显的重叠。将具有九种症状的患者人数与 DMD 患者人数的比例进行了比较。(比例 = OMD/DMD)。比例 >1 表示特定其他肌营养不良症 (OMD)中症状的发生率更高。OMD:其他肌营养不良症:DMD 杜氏肌营养不良症。
4.基因检测大数据分析结果解读
杜氏肌营养不良症 (DMD) 通常根据临床表现进行诊断,包括行走、站立和坐下困难、小腿肌肉肥大、跟腱早期挛缩、腿部肌肉无力、肌酸激酶水平升高、肌肉活检异常以及确认的分子分析。在肌营养不良基因检测的患者队列中,肌营养不良基因检测观察到了其他临床特征,例如足尖行走、频繁跌倒、Gower 征和蹒跚步态。另一方面,肢带型肌营养不良症 (LGMD) 是一类由 15 个或更多基因改变引起的多样化遗传疾病。这些基因改变会影响肌肉纤维,导致没有特定或独特的临床特征来预测多样化的基因型变异。由于临床表现相互重叠且有时十分复杂,临床医生很难仅根据患者症状做出准确的临床诊断。因此,LGMD 患者很容易被误认为 DMD/BMD 患者,尤其是后者症状较轻时。本报告强调了从基因上准确诊断肌营养不良症的必要性,并通过适当的咨询计划实施疾病管理和预防策略。
在 961 名先前描述的临床疑似 DMD 患者中,靶向下一代序列分析结合全外显子组测序 (WES) 显示,105/961 (10.9%) 患有与其他肌营养不良症相关的基因变异,包括 Bethlem 和 Ullrich 先天性肌病、Emery‐Driefuss 肌营养不良症和肢带型肌营养不良症 (LGMD)。这些肌营养不良症大多数被发现是常染色体隐性遗传病,其中包括具有纯合变异的患者以及少数具有复合杂合变异的患者。有趣的是,三分之一的其他肌营养不良症患者被发现患有 LGMD2E,这是一种严重的 LGMD 形式,会影响非常年幼的儿童。另外 34/961 (3.5%) 名患者被发现患有其他疾病,包括夏科-马里-图斯病和杆状体肌病等。一小部分患者 (6/961;0.6%) 尽管有 DMD 样症状,但仍未确诊,因为没有已知的其他肌营养不良症 (OMD)基因变异。有趣的是,这六名患者中有三名在首次出现症状时经常跌倒和行走困难。虽然三人中的两人在 28 岁和 14 岁时仍能行走,但一名患者在 10 岁时就无法行走了。这六个病例需要进一步检查,也许可以通过全基因组测序,以精确识别可能导致诊断其潜在疾病的基因变异。
在墨西哥进行的一项类似研究中,72 名临床怀疑患有肌营养不良症且没有DMD基因缺失证据的 18 岁以下无血缘关系男性,NGS 分析显示 68% 患有DMD基因变异,12.5% 患有常染色体隐性 LGMD 相关基因型,包括 LGMD 类型 2A-R1、2C-R5、2E-R4、2D-R3 和 2I-R9。肌营养不良基因检测临床大数据分析中没有 LGMD2B-R2 的原因是研究中纳入的患者数量较少。
CAPN3基因是第一个与肌营养不良症相关的非结构蛋白, 据报道该基因有 400 多个突变。然而,突变CAPN3在 LGMD2A 致病性中 的作用仍不清楚。众所周知,LGMD2A 是全球最常见的 LGMD 类型。在一名疑似 LGMD2A 患者中发现了一个有趣的现象(表 6,患者 #5)。虽然该患者CAPN3基因(外显子 18;c.2033A>T)的错义突变杂合子与 LGMD 2A 有关,但进一步研究发现CLCN1基因(外显子 18;c.1667T>A)存在第二个杂合突变,该突变与先天性肌强直(汤姆森病)有关。基于这一观察,该患者以及其他其他肌营养不良症 (OMD)相关基因存在多个突变的患者(甚至基因分析)的准确诊断仍不清楚。因此,需要注意的是,虽然一种或多种变异对所述肌营养不良症的贡献可能很大,但目前还无法就这些突变基因对疾病致病性的贡献得出任何结论。
在 11 名被确诊患有CAPN3变异的患者中, 由于两名无血缘关系的患者具有相同的 CAPN3 基因突变,因此患者 #6(9 岁)和 #7(6 个月)之间的发病年龄存在令人惊讶的显著差异(表6 ) 。进一步检查两名患者的突变谱后发现,虽然 #6 在CAPN3基因中发生纯合突变(外显子 18;c.2003T>G),但 #7 在 PLEC 基因中发现了另外两个杂合变异(外显子 28;c.4198G>A 和外显子 32;c.8887G>A)。这些额外的变异可能导致后者发病年龄更早,这表明与肌肉功能相关的基因的多种突变可能共同导致疾病的严重程度。
之前对 238 名欧洲 LGMD2A 患者的研究发现,发病平均年龄为 13.8±8.1 岁,范围为 2-49 岁。该研究发现 30.7% 的患者在CAPN3基因中存在 c.2362AG>TCATCT 插入突变。在肌营养不良基因检测的队列中未观察到此突变(表 6)。
LGMD2Q 的特征是近端肌肉无力,偶尔跌倒,爬楼梯困难,并且病程进展导致成年早期失去行走能力,这是由PLEC基因变异引起的 (Irwin McLean et al., 1996 )。在 7 名患有PLEC基因变异的其他肌营养不良症 (OMD)患者中,有 4 名被发现具有PLEC基因复合杂合变异。另一方面,3/7 患者具有PLEC基因杂合变异以及DYSF(外显子 34;c.3779G>A 和内含子 34;c.3898-4C>G)和LAMA2(外显子 14;c.1963C>T)基因的其他变异(表 8)。此外,患者 1 和 6 仅含有杂合 PLEC 基因变异。因此,对这些患者进行进一步分析可能有助于了解PLEC基因中的单个或多个变异,加上与其他肌营养不良症 (OMD)相关的基因中的其他变异是否会导致疾病。
肌营养不良基因检测队列中三分之一的常染色体隐性肌营养不良症患者被发现具有与 LGMD2E 相关的SGCB基因变异。虽然 LGMD2E 的临床症状各不相同,但诊断年龄通常在 10 岁以下。在十几岁中后期,患者会丧失行走能力。肌营养不良基因检测队列中的 29 名 LGMD2E 患者中,近三分之二是近亲结婚的产物。有趣的是,29 名无血缘关系的患者中有 18 名都属于泰米尔纳德邦,他们在SGCB基因的外显子 4 中携带相同的 c.544A>C 变异,这可能会破坏肌营养不良蛋白复合体的完整性。在 8 个伊朗家族中也观察到了类似的结果,这些家族具有相同的单倍型,其SGCB基因发生致病突变,导致外显子 2 缺失,从而形成 β‐肌聚糖蛋白的跨膜结构域。这项研究还提出了奠基者效应(Mojbafan et al., 2020 )。在第二项来自伊朗的研究中,Alavi 等人表明,14 名 LGMD2E 患者中有 12 名发生缺失突变,导致SGCB基因外显子 2 丢失。有趣的是,12 名出现这种缺失的患者中有 10 名来自伊朗南部和东南部,基于三个单核苷酸多态性 (SNP) 标记的单倍型分析强烈表明存在奠基者效应的可能性)。在肌营养不良基因检测队列中,来自印度泰米尔纳德邦同一地理区域的多名无亲属患者中发现SGCB基因的相同突变,这提示存在奠基者效应。要确认这一发现,需要更多患者样本和进一步分析。这是首次报道印度 LGMD2E 患者中存在多名 c.544A>C 点变异。
此前已有研究表明,LGMD2E 是 LGMD 中最严重的类型之一。肌营养不良基因检测患者队列的数据证实了这一点,并进一步表明,当比较发病年龄和丧失行走能力的年龄时,LGMD2E 和 DMD 是相当的。这一观察结果或许可以解释为什么肌营养不良基因检测的队列中有大量 LGMD2E 患者被误认为是 DMD 患者。
肌营养不良基因检测的分析明确表明,不建议仅根据症状对患者进行诊断,因为这可能会导致多达 15% 的病例被误诊为 DMD,正如肌营养不良基因检测在队列中观察到的那样。因此,强烈建议使用明确定义的肌营养不良症组通过 NGS 进行基因分析,并应将其纳入肌营养不良基因检测之前推荐的诊断工作流程(图 1)中,用于出现 DMD 样症状的患者。使用这种方法的结果可能会影响治疗以及患者及其家属的咨询,从而对患者的健康起着至关重要的作用。
肌营养不良基因检测对印度泰米尔纳德邦 LGMD 和其他常染色体隐性遗传病患病率的研究不应得出任何流行病学结论,主要是因为肌营养不良基因检测的研究最初旨在识别和诊断 DMD 患者,因此肌营养不良基因检测临床大数据分析仅考虑了男性。
通过对 961 名临床疑似 DMD 患者进行完整的基因分析,肌营养不良基因检测的研究结果表明,仅根据临床标准,大约 15% 的患者可能会被误诊为患有 DMD。这项研究强调了进行完整的基因检查以准确诊断肌营养不良症和肌病患者的必要性。因此,疑似肌营养不良症病例,尤其是那些疑似患有 DMD 的病例,如果在 mPCR 和 MLPA 分析后没有得到确认的分子诊断,则需要通过下一代测序进行调查。这项工作将确认肌营养不良症的类型,以便尽早启动适当的咨询计划,以告知预防策略并启动治疗方案(如果有)。这些全面的努力旨在提高患者的生活质量,并赋予携带者女性(如适用)和近亲结婚的父母权力,使他们能够做出明智的生育选择,以阻止肌营养不良症在社区中的传播。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
