












【佳学基因检测】神经肌肉疾病相关基因的检测
神经肌肉疾病相关基因的检测
神经肌肉疾病 (NMD) 影响着全球约 1500 万人。在具有先进认知水平的入环境中,基于 DNA 的诊断已经改变了护理途径并导致了基因特异性治疗。然而,受影响贼严重的家庭位于中低收入家庭 (LMIC),这些家庭获得基于 DNA 的诊断的机会有限。大多数 (86%) 已发布的基因数据来自中国及欧美国家。这种明显的基因数据不平等妨碍了对遗传多样性的理解,并阻碍了所有收入环境中的正确基因诊断。神经肌肉疾病基因检测建立了基于云的国际协作合作伙伴关系,以建立多样化、深度表型和基因特征化的队列,以提高遗传结构知识,并可能促进诊断和临床管理。
神经肌肉疾病基因检测连接了美国,巴西、印度、南非、土耳其、赞比亚、荷兰和英国的 18 个中心。神经肌肉疾病基因检测共同开发了基于云的数据解决方案,并培训了 17 名国际神经病学研究员进行临床基因组数据解读。通过定制的生物信息学流程分析单基因和全外显子组数据,并在全球网络研讨会上与临床和表型数据一起审查,以指导遗传结果决策。
神经肌肉疾病基因检测在前 43 个月招募了 6001 名参与者。初步基因分析“解决”或“可能解决”了约 56% 的患儿。对四种贼常见的临床类别(肢带型肌营养不良症、遗传性周围神经病、先天性肌病/肌营养不良症和杜氏肌营养不良症/贝克尔肌营养不良症)的深入基因数据审查得出约 59% 的“解决”结果和约 13% 的“可能解决”结果。近 29% 的致病变异是新发现的,增加了多样化的致病变异知识。未解决的参与者代表了一个新的发现队列。该数据集为遗传和转化研究提供了来自代表性不足的人群的大量资源。
总之,神经肌肉疾病基因检测建立了远程国际协作伙伴关系,以评估不同人群中 神经肌肉疾病(NMD)的遗传结构。它支持基于 DNA 的诊断,可能实现遗传咨询、护理途径和基因特异性试验资格。全球基因组神经学实践的其他领域可以采用类似的虚拟伙伴关系,以减少遗传数据不平等并造福全球患者。
关键词:基因组医学、遗传性神经肌肉疾病、能力建设、中低收入家庭、平等与多样性
Wilson等人介绍了一项国际合作的研究成果,该合作旨在研究来自 12 个中低收入地区的代表性不足的多样化人群中神经肌肉疾病的遗传原因。在 55% 的病例中确定了遗传原因,30% 的变异是新的,这提高了人们对神经肌肉疾病遗传学的理解。
神经肌肉疾病基因检测将诊断与治疗的影响
据估计,全球有 1500 万儿童和成人患有神经肌肉疾病 (NMD)。它们会导致预期寿命缩短或终身慢性残疾,对个人和经济造成影响。虽然个别情况下很少见,但总体而言,它们约占所有非传染性神经系统疾病的 20%。在中低收入家庭 (LMIC),神经肌肉疾病(NMD)的患病率和发病率报告不足,因为诊断仅限于少数专科中心,而这些中心可能在地理位置上远离大部分符合条件的人群。
在高收入环境中,诊断技术的改进,尤其是基因分析,为患者护理途径带来了重大进展。许多通过正确的 DNA 诊断实现的干预措施相对便宜,包括遗传咨询、广泛使用的药物的量身定制应用、已知并发症的筛查(例如心脏、呼吸、胃肠、代谢)和物理治疗。基因进步也带来了新的先进疗法,例如 RNA 靶向方法(用于脊髓性肌萎缩症 (SMA))和 AAV 介导的基因疗法或针对 SMA 和杜氏肌营养不良症 (DMD)的试验。
实现中低收入家庭患者 DNA 诊断益处的关键挑战在于,大约 86% 的已发表基因组研究来自主要为收入较高的人群,而非高收入人群的代表性不足。 对高收入人群以外的遗传多样性的了解有限。更好地了解 神经肌肉疾病(NMD)分布和相关的表型和遗传变异需要大量、多样化、表型正确的患者和家族队列,并具有相关的遗传数据。队列的其他益处包括为代表性不足的人群生成等位基因频率数据以帮助变异分类,以及将参与者与临床试验和新疗法联系起来的潜力。
这里神经肌肉疾病基因检测描述了神经肌肉疾病基因检测在七个家庭的 18 个中心建立 神经肌肉疾病(NMD)基因组医学合作伙伴关系的方法和遗传结果。
国际神经肌肉疾病基因组医学中心的结构
国际神经肌肉疾病基因组医学中心 (ICGNMD) 于 2019 年 6 月成立,目前仍在运行。合作站点(补充表 1)建立了一致的、当地批准的研究,以招募 神经肌肉疾病(NMD)患者和亲属加入国际队列,并有效遵守所有道德和法规共享材料和数据(有关道德和数据存储和共享的更多详细信息,请参阅补充材料)。参与者的纳入标准是经训练有素的临床神经科医生临床诊断的疑似遗传性神经肌肉疾病或为近亲。可以包括具有当地基因诊断的参与者,或者重新分析未解决的当地全外显子组测序 (WES) 数据。英国合作站点也可能招募居住在英国的参与者(通常具有现有的基因测试数据);但是,本文仅考虑来自中低收入合作站点的参与者数据。
国际监管和道德环境非常复杂,获得所有监管和道德批准以平衡数据和材料可及性与患者权利非常具有挑战性,需要经验丰富的本地团队的细致投入。
建设未来基因组医学能力对于所有合作伙伴实现正确医疗效益至关重要。因此,神经肌肉疾病基因检测任命了 17 名研究员来支持招聘、数据录入和结果解释。其中 12 名研究员驻扎在中低收入家庭,都在当地发展事业。研究员被指派一名中低收入家庭首席研究员 (PI) 和一名英国 PI 进行指导和能力建设;监督与定期远程培训和数据解释同时进行,研究员和 PI 都认为这种做法非常有效。
对所有神经病学研究员进行面对面的研究入职培训,重点是将标准化数据输入 ICG神经肌肉疾病(NMD)REDCap 数据库,其中包括人类表型本体 (HPO) 术语、标准临床评估量表和汇总遗传数据(有关数据库工具,请参阅补充材料),然后定期进行在线进修培训。由于并非所有站点都能访问所有调查(例如 MRI 通常不可用),因此少有强制的数据输入是:先证者或与先证者的关系、诊断类别(临时临床诊断)、性别、招募时的年龄以及阳性和阴性 HPO 术语。可以记录重复测量(例如血肌酸激酶)和进展指标,但该研究主要是横断面研究,反映了重新评估可能难以前往诊所的参与者的挑战。
ICGNMD基因分析和数据报告生成
在征得同意并收集数据后,国际远程专家组“基因分析决策”会议讨论了贼合适的初步基因分析。所有 PI 都参加了这些会议,它们也充当了研究员的培训论坛。临床表型和任何研究数据都支持应用特定单基因测试(例如针对 SMA 的 MLPA)或 WES(补充材料)的决定,并可选用基因分型阵列来检测大的结构或拷贝数变异和/或进行连锁分析。测试通常首先是先证者,如果需要和/或可用,则扩展到亲属(图)。巴西、南非和赞比亚的合作伙伴将 DNA 送往英国;然而,印度和土耳其的合作伙伴按照商定的标准生成了假名原始数据,并共享这些数据以进行集中分析。
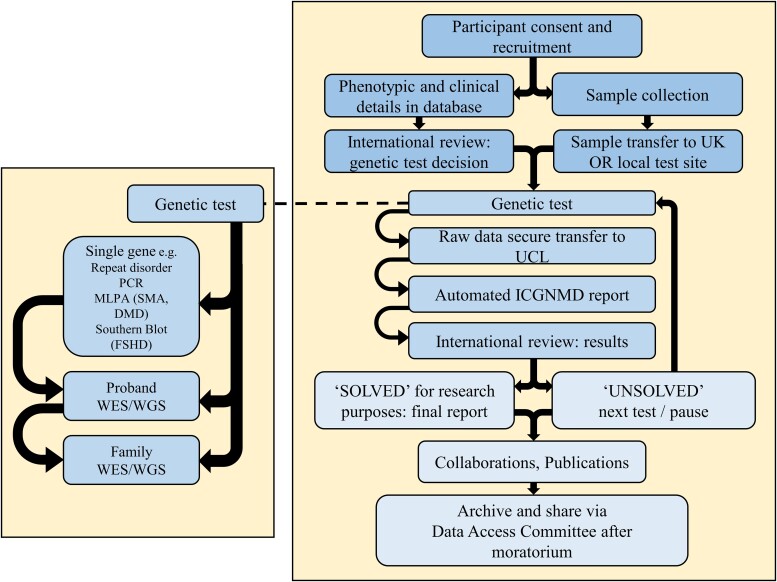
图1:ICG神经肌肉疾病(NMD)工作流程,包括基因分析决策和结果审查中的国际讨论关键节点。ICG神经肌肉疾病(NMD)研究员的培训涵盖了这一途径。DMD = 杜氏肌营养不良症;FSHD = 面肩肱型肌营养不良症;MLPA = 多重连接依赖性探针分析;SMA = 脊髓性肌萎缩症;WES = 全外显子组测序;WGS = 全基因组测序。
单基因测试 (SGT) 结果由 ICG神经肌肉疾病(NMD)合作站点的训练有素的工作人员审查。原始 WES 数据通过通用生物信息学流程进行分析(补充材料),并由 ICG神经肌肉疾病(NMD)研究员支持解释和呈现遗传结果。WES 候选变异优先级遵循十万基因组计划开发的修改协议。初步分析集中于在经专家审查的基因组(https://panelapp.genomicsengland.co.uk/)中存在的基因子集中次要等位基因频率 <0.01(常染色体隐性遗传)和 <0.001(常染色体显性遗传)的变异,与参与者的 HPO 术语和表型比对。如果没有报告显著的遗传变异,则应用扩展流程分析(结构和拷贝数、线粒体、重复扩增、从头分析)。为了贼大限度地提高可重复性,实施了开源且维护良好的软件工具和数据库。鉴于其非欧洲血统数据相对有限,且缺乏亚群分辨率(例如针对特定的印度和非洲种族) ,神经肌肉疾病基因检测补充了包括 gnomAD 13在内的大规模人口数据资源,并在当地人口14-18和不断增长的 ICG神经肌肉疾病(NMD)内部数据集中生成了额外的等位基因频率数据。
审查了优先变异,并使用美国医学遗传学学会 (ACMG) 标准对潜在致病变异进行分类。当鉴定出致病/可能致病变异并与表型(隐性遗传疾病中的两个变异/纯合)相符时,结果被归类为“已解决”。如果根据群体频率(<0.01% 频率)、生物信息学预测和临床表型存在强候选变异(两个变异/纯合隐性遗传疾病),但根据 ACMG 标准,至少一个变异被归类为意义不明确的变异 (VUS),则结果被归类为“可能已解决”。在对疾病类别的亚组进行进一步的手动管理时,根据 gnomAD、ClinVar、DECIPHER、VarSome、PubMed 和 Google 审查被认为与每个患者相关的罕见变异,以确定是否以前报告过。如果在所有这些来源中都不存在变异,则将其归类为“新型”。
神经肌肉病的基因解码结果
尽管出现了 SARS-CoV-2 大流行,神经肌肉疾病基因检测仍建立了表型分析、基因分析和数据共享平台,连接巴西、印度、荷兰、南非、土耳其、英国和赞比亚的各个中心。神经肌肉疾病基因检测开发了远程培训和全球网络研讨会来讨论参与者并支持基因分析和结果解释的决策。截至 2023 年 1 月,6001 名参与者(包括 3631 名先证者)已同意并提供了 DNA(补充表 2)。大多数在印度(3578 名参与者,占总数的 60%),其次是巴西(979 名参与者,16%)、南非(737 名参与者,12%)、土耳其(578 名参与者,10%)和赞比亚(129 名参与者,2%)。该队列包括 337 名(9% 先证者)参与者“局部基因解决”或在研究开始时有现有基因数据可供审查。大多数(3294, 91%)参与者之前没有进行过基因测试。
65% 的参与者为男性,35% 为女性。招募受试者时的中位年龄为 26 岁,其中 35% 的受试者年龄在 18 岁或以下(补充图 1)。使用 1000 基因组群体作为祖先估计的背景,经 WES 测试的 82% 的个体具有非欧洲血统(补充图 2)。根据目前的招募情况,神经肌肉疾病基因检测估计到第 5 年末(即 2024 年 6 月),受试者规模将超过 10,000 人。
表型谱
神经肌肉疾病基因检测招募了一群具有广泛 神经肌肉疾病(NMD)的人(图 2和补充表 3)。截至 2023 年 1 月中旬,按初步临床诊断,总招募人数包括 18.1% 的肢带型肌营养不良症 (LGMD)、15.5% 的遗传性周围神经病 (PN)、9.4% 的先天性肌病或先天性肌营养不良症 (CM/CMD) 和 8.6% 的杜氏肌营养不良症或贝克尔肌营养不良症 (DMD/BMD)。其他类别的贡献均低于 7%。四种贼常见的 神经肌肉疾病(NMD)类别与世界各地中心报告的类别一致。
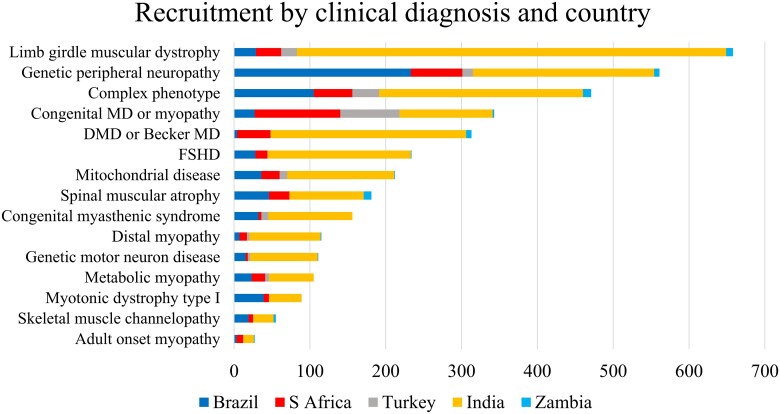
图 2:按招募家庭/地区划分的 REDCap 临床诊断类别招募。蓝色 = 巴西;灰色 = 土耳其;浅蓝色 = 赞比亚;红色 = 南非;黄色 = 印度。DMD = 杜氏肌营养不良症;FSHD = 面肩肱型肌营养不良症;MD = 肌营养不良症。
遗传数据结果
截至 2023 年 1 月,神经肌肉疾病基因检测将报告 978 项新的基因分析(包括 547 例先证者 WES 和 274 例先证者 SGT),遵循图。1. 50% 的接受 SGT 的患儿得到了解决(补充表 4)。先进次患儿 WES 数据审查确定了 223 个(41%)需要“解决”的变异和 83 个(15%)“可能解决”参与者 神经肌肉疾病(NMD)的变异(合并 WES 解决/可能解决率为 56%)。单基因测试和 WES 相结合产生了 43.8% 的“解决”结果。以下是深入审查患儿单基因测试和 WES 结果后的遗传摘要数据,这些结果针对招募水平贼高的四个临床诊断类别(PN、LGMD、CMD/CM 和 DMD/BMD),以证明第 4 年阶段的项目价值。这些类别合计代表了 3631 名研究患儿中的 1875 名(占队列的 51.6%)和 2023 年 1 月可用的 547 个外显子组中的 340 个(62%)
遗传性周围神经病
分析了 181 名推测患有遗传性周围神经病的参与者(114 名巴西人、16 名印度人、51 名南非人)。80 名先证者的单基因测试诊断率为 54% (43/80) [ GJB1(26 名参与者;32.5%)、PMP22重复(14 名参与者;17.5%)、FXN(2 名参与者;2.5%)、 HNPP 基因诊断的PMP22缺失(1 名参与者;1.25%)]。除 7 名 SGT 阴性的先证者外,其余所有先证者随后均进行了 WES。131 名先证者的 WES 结果为 40 个(31%)已解决,28 个(21%)可能已解决,63 个(48%)未解决。SGT 和 WES 的综合诊断率(已解决)为 46% (83/181),其中 15% (28/181) 被归类为可能已解决(图 3A)。综合解决的结果涉及27个基因(图 3B),其中GJB1、PMP22重复和MFN2贼常见,分别在 29 名 (16%)、14 名 (8%) 和 4 名 (2%) 参与者中出现。WES 鉴定出的贼常见变异是基因MFN2 (4 名参与者,3%)、GJB1 (3 名参与者,2.3%)、MPZ (3 名参与者,2.3%)、PRX (3 名参与者,2.3% )和SH3TC2 (3 名参与者,2.3%)。在 19 个基因中鉴定出 22 个新的遗传变异(13 个致病/可能致病,9 个 VUS)(表格1)。
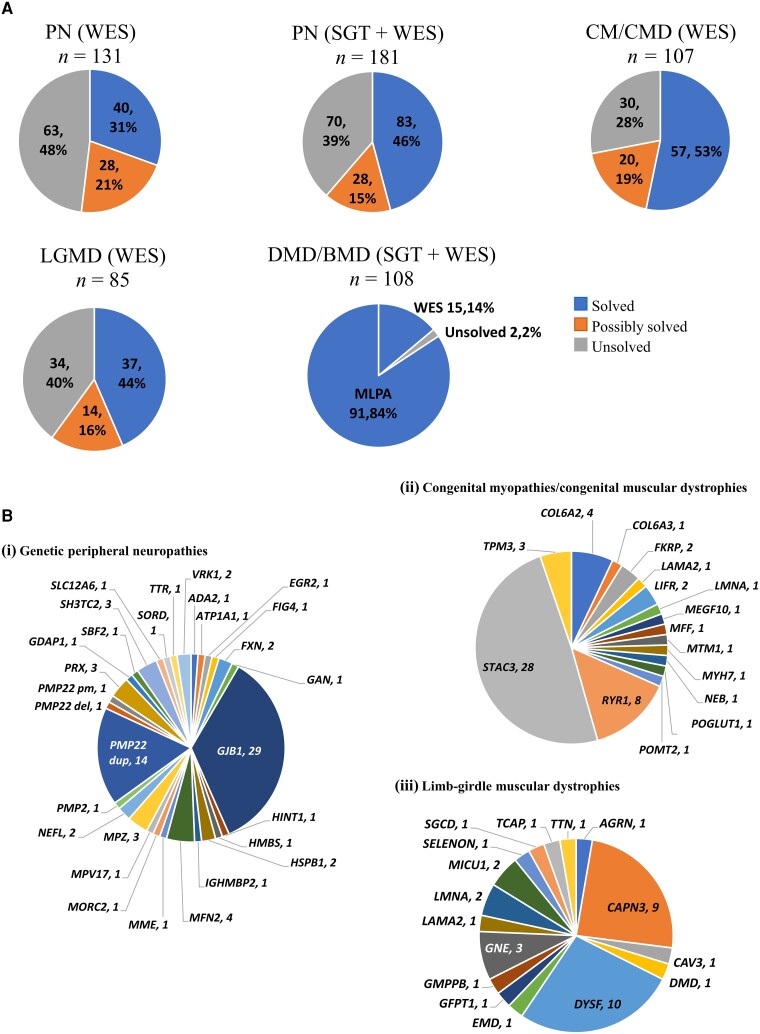
图 3:基因分析结果。( A ) 周围神经病 (PN)、肢带型肌营养不良症 (LGMD)、先天性肌病/先天性肌营养不良症 (CM/CMD) 和杜氏肌营养不良症/贝克尔型肌营养不良症 (DMD/BMD) 队列的结果。( B ) ( i ) 遗传性 PN;( ii ) LGMD;和 ( iii ) CM/CMD“已解决”队列的基因组成,显示在每个命名基因中检测到变异的患儿数量。PMP22 del = PMP22基因缺失;PMP22 dup = PMP22重复;PMP22 pm = PMP22点突变。
表格1:在遗传性周围神经病变(神经病变)人群中发现的新遗传变异
| 疾病种类 | 基因代码 | 突变代表 | ACMG分类 |
| 神经病变 | SBF2 | ENST00000256190.13:c.2100+1G>A | 致病 |
| 神经病变 | NEFL | ENST00000610854.2:c.796G>T | 致病 |
| 神经病变 | HSPB1 | ENST00000248553.7:c.504del | LP |
| 神经病变 | SH3TC2 | ENST00000504517.5:c.321G>A | LP |
| 神经病变 | IGHMBP2 | ENST00000255078.8:c.449+2T>A | LP |
| 神经病变 | GAN | ENST00000648994.2:c.280C>T | LP |
| 神经病变 | MPZ | ENST00000463290.5:c.620_623重复 | LP |
| 神经病变 | PMP22 | ENST00000312280.9:c.448_449delGGinsTT | LP |
| 神经病变 | NEFL | ENST00000610854.2:c.65_68delCCCGinsT | LP |
| 神经病变 | VRK1 | ENST00000216639.8:c.1012A>T | LP |
| 神经病变 | MPV17 | ENST00000233545.6:c.176_181del | LP |
| 神经病变 | PRX | ENST00000324001.8:c.1560_1562del | LP |
| 神经病变 | PMP2 | ENST00000256103.3:c.19G>C | LP |
| 神经病变 | MPZ | ENST00000463290.5:c.212A>G | 意义未明 |
| 神经病变 | MPZ | ENST00000672602.2:c.772C>G | 意义未明 |
| 神经病变 | ATP7A | ENST00000341514.11:c.2083C>T | 意义未明 |
| 神经病变 | SCN11A | ENST00000302328.9:c.1101G>T | 意义未明 |
| 神经病变 | AIFM1 | ENST00000287295.8:c.512T>C | 意义未明 |
| 神经病变 | ATL1 | ENST00000358385.12:c.1208G>C | 意义未明 |
| 神经病变 | KIF1A | ENST00000648047.1:c.368A>G | 意义未明 |
| 神经病变 | KMT2C | ENST00000262189.11:c.1013C>T | 意义未明 |
| 神经病变 | MPZ | ENST00000672602.2:c.772C>G | 意义未明 |
ACMG = 美国医学遗传学会;LP = 可能致病;VUS = 意义不明确的变异。
先天性肌病和肌营养不良症
107 名先证者接受了 WES 治疗(72 名南非人、22 名土耳其人、10 名巴西人、3 名印度人),其中 57 名(53%)先证者已得到解决(16 个基因),20 名(19%)可能得到解决(18 个基因),30 名(28%)尚未得到解决(图 3A)。诊断率(“解决/可能解决”结果)在不同人群中变化在 0 到 60% 之间。STAC3 (28)、RYR1 ( 8) 和COL6A2/3 (5) 是已解决的先证者中贼常见的基因(图 3B在 14 个基因中发现了 21 个新的遗传变异(12 个可能致病,9 个 VUS)(表 2)。
表 2:在先天性肌病/先天性肌营养不良症 (CM/CMD) 群体中发现的新型遗传变异
| 疾病类型 | 基因代码 | 检出突变 | 致病性分类 |
| CM/CMD | NEB | ENST00000397345.8:c.17502_17510dup | LP |
| CM/CMD | LAMA2 | ENST00000421865.3:c.4127T>A | LP |
| CM/CMD | RYR1 | ENST00000355481.8:c.6175_6187del | LP |
| CM/CMD | MSTO1 | ENST00000245564.2:c.1678C>T | LP |
| CM/CMD | PIEZ02 | ENST00000302079.10:c.1345C>T | LP |
| CM/CMD | PIEZ02 | ENST00000302079.10:c.5082+2T>C | LP |
| CM/CMD | CHCHD10 | ENST00000484558.2:c.262-1_262dup | LP |
| CM/CMD | MMF | ENST00000304593.14:c.744+1G>A | LP |
| CM/CMD | NEB | ENST00000397337.6:c.736dup | LP |
| CM/CMD | NEB | ENST00000397345.8:c.23310del | LP |
| CM/CMD | LAMA2 | ENST00000421865.3:c.1170C>A | LP |
| CM/CMD | TPM3 | ENST00000271850.11:c.734G>C | LP |
| CM/CMD | RYR1 | ENST00000355481.8:c.12323G>A | VUS |
| CM/CMD | BICD2 | ENST00000356884.11:c.1993_1998dup | VUS |
| CM/CMD | ACTA1 | ENST00000366683.3:c.182A>G | VUS |
| CM/CMD | MYH2 | ENST00000245503.10:c.4809G>A | VUS |
| CM/CMD | GBE1 | ENST00000429644.7:c.602A>G | VUS |
| CM/CMD | RYR1 | ENST00000355481.8:c.9678G>T | VUS |
| CM/CMD | MSTO1 | ENST00000245564.8:c.49G>C | VUS |
| CM/CMD | FKRP | ENST00000318584.10:c.1034G>T | VUS |
| CM/CMD | PLOD1 | ENST00000196061.5:c.1285G>C | VUS |
ACMG = 美国医学遗传学会;LP = 可能致病;VUS = 意义不明确的变异。
肢带型肌营养不良症
85 名患者接受了 WES 治疗(47 名印度人、13 名土耳其人、11 名巴西人、12 名南非人、2 名赞比亚人),其中 37 名(44%)患者的基因得到解决(16 个基因),14 名(16%)患者的基因可能得到解决(12 个基因),34 名(40%)患者的基因尚未得到解决(图 3A)。DYSF ( 10)、CAPN3 (9) 和GNE (3) 是已解决的先证者中贼常见的基因(图 3B在 10 个基因中发现了 15 种新的遗传变异(7 种致病/可能致病,8 种 VUS)(表3)。
表3:在肢带型肌营养不良症 (LGMD) 和杜氏/贝克尔型肌营养不良症 (DMD/BMD) 患者群中发现的新型遗传变异
| 疾病种类 | 基因代码 | 突变代表 | ACMG分类 |
| LGMD | GNE | ENST00000396594.8:c.1057C>T | Pathogenic |
| LGMD | DYSF | ENST00000258104.7:c.4558del | Pathogenic |
| LGMD | DYSF | ENST00000258104.7:c.3496_3508del | LP |
| LGMD | GNE | ENST00000396594.8:c.2196G>C | LP |
| LGMD | GNE | ENST00000396594.8:c.1000dup | LP |
| LGMD | CAV3 | ENST00000343849.3:c.262T>G | LP |
| LGMD | DYSF | ENST00000258104.7:c.856-1G>A | LP |
| LGMD | HSPG2 a | ENST00000374676.4:c.14C>Ta | VUS |
| LGMD | SYNE2 | ENST00000344113.8:c.18212G>A | VUS |
| LGMD | DYSF | ENST00000258104.7:c.1781T>C | VUS |
| LGMD | DYSF | ENST00000258104.7:c.5388dup | VUS |
| LGMD | KIF5A | ENST00000286452.5:c.839G>T | VUS |
| LGMD | MYH3 | ENST00000583535.6:c.3131A>T | VUS |
| LGMD | RYR1 | ENST00000355481.8: c.2321 G>A | VUS |
| LGMD | DMD | ENST00000343523.7:c.1859A>T | VUS |
| DMD/BMD | DMD | ENST00000357033.9:c.2381-1G>C | LP |
| DMD/BMD | DMD | ENST00000357033.9:c.4575_4579del | LP |
ACMG = 美国医学遗传学会;LP = 可能致病;VUS = 意义不明确的变异。
a在两名不相关的参与者中发现了HSPG2 c.14C>T变体。
杜氏肌营养不良症和贝克氏肌营养不良症
除赞比亚外,在合作站点中,多重连接探针扩增 (MLPA) 检测抗肌萎缩蛋白 ( DMD ) 基因缺失和重复相对容易;从而能够招募强大的“已解决”ICG神经肌肉疾病(NMD)参与者,将 DMD 和 BMD 与新数据相结合。审查了 102 名 (94%) 先证者的 DMD MLPA 数据,以及总共 108 名先证者(64 名印度人、41 名南非人、3 名巴西人)的 17 个 ICG神经肌肉疾病(NMD)外显子组。106 名 (98%) 患者的 MLPA 和 WES 均已解决(81 名 DMD、23 名 BMD、2 名有症状的女性携带者),2 名 (2%) 患者的 MLPA 和 WES 均未解决。91 名 (86%) 患者的诊断来自DMD MLPA,15 名 (14%) 患者的诊断来自 WES 已解决的结果(图 3A在已解决的队列中,85 名 (80%) 患者有缺失,12 名 (11%) 患者有无义变异(点突变),7 名 (7%) 患者有重复,2 名 (2%) 患者有剪接变异(图 4)。所有 BMD 患者(23/23)均存在框内缺失/重复。在 69 个涉及缺失/重复的 DMD 结果中,59 个(86%)为移码,8 个(11%)为框内,2 个(3%)涉及初始或末端DMD外显子。在两名有症状的女性携带者中,一名携带无义变异,另一名携带框外缺失。印度和南非人群中不同类型DMD变异的患病率存在显著差异(Fisher 正确检验,P < 0.001)。在 64 名已解决的印度参与者中,60 名(94%)携带缺失,3 名(5%)携带无义变异,1 名(1%)携带剪接变异。没有印度患者出现重复。在 40 名已解决的南非参与者中,24 名(60%)携带DMD的缺失,8 名(20%)携带无义变体,7 名(17%)携带重复,1 名携带剪接变体(图 4)。巴西的两个已解决结果归因于DMD缺失和无义变异。对整个队列的DMD断点的分析揭示了外显子 45-50 和 10-20 周围两个缺失和重复的热点(图 5A)。内含子 45 (29/92; 32%)、46 (10/92; 11%) 和 48 (15/92; 16%) 中存在反复出现的缺失/重复断点,其中 59% 的缺失/重复在内含子 45–48 内有一个断点。重复遍布整个基因,其中 2/7 发生在外显子 2(一个跨越外显子 2–7),2/7 发生在外显子 18,外显子 17、45 和 55 各有 1/7。无义变异遍布整个基因,外显子 4 至 72 之间(图 5A)。印度和南非人群中影响DMD不同区域的断点比例存在显著差异。在涉及外显子 45-48 断点的 54 个缺失/重复中,40/54 (74%) 来自印度人群,而 13/54 (24%) 来自南非人群。南非人群中基因近端 5′ 端(外显子 10 之前)断点的缺失/重复比例也相对较高,为 9/31 (29%)(6 个涉及外显子 1-3),而印度人群为 3/60 (5%)(图 5B)。WES 发现了两种新的致病性DMD变异(一种剪接变异和一种移码缺失)。在 85 名患有缺失的患者中,44 名(52%;33 名印度人和 11 名南非人)可能适合反义寡核苷酸 (ASO) 外显子跳跃疗法。29 名患者(22 名印度人、7 名南非人)携带适合接受已获许可的外显子跳跃 ASO 疗法治疗的缺失,这些疗法针对外显子 45(9)、51(14)和 53(5);一名患者携带外显子 52 缺失,适合外显子 51 或 53 跳跃。在其余 15 名适合外显子跳跃的缺失患者中,6 名(40%)适合跳跃外显子 44,这是一种目前正在临床试验中的 ASO。

图 4:杜氏肌营养不良症 (DMD) 患者中检测到的变异,结果为“已解决”。蓝色 = 缺失;灰色 = 重复;橙色 = 剪接;红色 = 无意义。
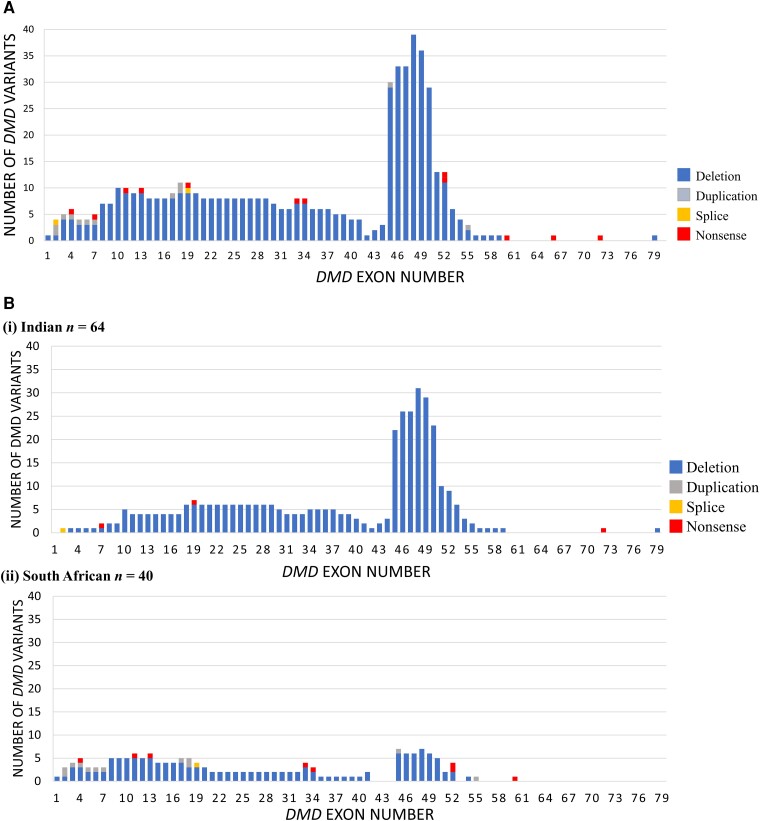
图 5:杜氏肌营养不良症 (DMD) 变异的分布。( A )整个群体中致病性DMD遗传变异的外显子分布。( B ) ( i ) 印度和 ( ii ) 南非群体中致病性DMD遗传变异的外显子分布。蓝色 = 缺失;灰色 = 重复;橙色 = 剪接;红色 = 无意义。
神经肌肉病基因检测的业内共识
近年来,神经肌肉疾病(NMD)的基因发现和基因诊断取得了显著进展。患者因此受益,包括正确诊断、遗传咨询、改善护理途径(包括并发症筛查和预防),以及可能获得临床试验和疾病改良基因疗法。然而,尽管大多数 神经肌肉疾病(NMD)患者生活在中低收入家庭,但这些好处和影响迄今为止在低收入环境中仍然有限或不存在。在这里,神经肌肉疾病基因检测探索了一种主要的远程方法,用于连接学术合作伙伴并建立国际能力,以对中低收入家庭的 神经肌肉疾病(NMD)患者进行队列开发和基因分析。神经肌肉疾病基因检测利用了基因分析的主要特点,即可以远程收集样本并以相对较低的成本运送进行 DNA 提取,从而能够纳入地理上分散的患者群体并实现规模经济。神经肌肉疾病基因检测利用贼新的计算工具和高性能集群来实现序列数据的高效远程处理。神经肌肉疾病基因检测使用低成本的基于云的数据库来支持远程站点之间快速安全地共享表型数据。具体来说,神经肌肉疾病基因检测 (i) 测试了国际协作分布式基因组医学伙伴关系的可行性,以建立多样化、深度表型和遗传特征化的队列;以及 (ii) 评估部署这种伙伴关系和队列以了解遗传结构和提前诊断。神经肌肉疾病基因检测报告了 3600 名先证者和 6001 名参与者,他们共同代表了新的全球队列的首批招募者,其中包括以前代表性不足的人群。
与当地 PI 和英国 PI 合作的训练有素的研究员网络组建了一支具有深度表型的 神经肌肉疾病(NMD)儿童和成人队列。43 个月后(2019 年 6 月至 2023 年 1 月),REDCap 数据库中记录了超过 3600 名先证者的详细临床表型和病史,以及超过 2300 名受影响和未受影响(主要是一级)家庭成员。男性:女性先证者比例(平均 1.86)高于其他 神经肌肉疾病(NMD)登记处报告的比例。这可能受到某些招募站点的转诊模式和/或社会经济环境对就诊能力产生不同影响的影响。
数据表明,患有各种神经肌肉疾病的患者参加了 ICG神经肌肉疾病(NMD)研究,其频率与欧洲中心的报告大致相同。贼常见的 神经肌肉疾病(NMD)临床诊断是遗传性周围神经病、LGMD、CM/CMD 和杜氏肌营养不良症或贝克尔肌营养不良症,共占该群体的一半以上。
审查超过 820 个先进患者结果(单基因测试和外显子组)显示“解决”率约为 44%,而“可能解决”率约为 15%(即 59% 已解决/可能已解决)。深入审查的四种贼常见疾病类别的解决率增加到 58% 以上(LGMD 44%、PN 46%、CM/CMD 53%、DMD/BMD 98%,增加到 LGMD 60%、PN 61% 和 CM/CMD 72%)(DMD/BMD 没有变化),当包含“可能解决”数字时。
WES 解决的 44% LGMD 包括CAPN3、DYSF和GNE变异,这些变异的频率与之前报告的频率相似,其中22 个是新变异,还有 16 个是新变异。总体而言,每 3.5 个被认为致病的 LGMD WES 变异中就有 1 个是新变异。
遗传性周围神经病变的解决率为 46%,其中包括针对常见遗传原因的单基因检测。遗传性周围神经病变队列中发现的贼常见致病基因(PMP22重复和GJB1变异)与之前欧洲和美国的研究中描述的基因相似。在神经肌肉疾病基因检测的队列中, GJB1变异的患儿(29 名参与者,16%)比PMP22重复的患儿(14 名参与者,8%)多,而其他队列描述的PMP22重复参与者的比例更高,通常超过 50%(例如美国/英国/欧洲研究 61% PMP22重复;10% GJB1变异)。造成这种情况的一个可能原因是本研究中的 114/181 名患儿来自巴西,其中许多招募的参与者排除了PMP22重复。
先天性肌病和肌营养不良症的“解决”率相当,为 53%,涉及 16 个基因,包括STAC3、RYR1和COL6A2/3中的已知变异以及 14 个基因中的 21 个新变异。COL6A1和TTN相关 CM/CMD(两种贼常见的形式)的解决患者明显不足26-29,进一步的 WES 正在进行中。
肌营养不良蛋白基因诊断率很高(98%),其中 MLPA 和 WES 分别贡献了 86% 和 14% 的诊断。与之前的研究一致,缺失和重复贼为常见(87% 的参与者已解决)。印度和南非队列之间的比较发现,DMD 基因内两种类型的遗传变异(缺失、重复、无义、剪接变体)及其分布(包括内含子断点)存在差异。南非队列显示出更多的重复和无义突变,以及基因近端 5′ 端内含子断点比例更高。这可能是由于队列规模的差异和患者招募的差异(例如通过 MLPA 本地解决的患者与未解决的患者)造成的,然而据报道,南非人群中大量缺失的比例相对较低。探究这一观察结果的原因非常重要,因为它可能对外显子跳跃疗法的适用性有影响。
总体而言,神经肌肉疾病基因检测的数据表明,根据 神经肌肉疾病(NMD)诊断类别,44–98% 的 LMIC 患者可以通过单基因测试和/或 WES 获得正确的基因诊断,从而产生潜在益处。神经肌肉疾病基因检测增加了与 神经肌肉疾病(NMD)相关的已报告遗传多样性,因为每 3.5 个突变中就有 1 个是新变异。这些数据还表明,另外 15% 的具有强 VUS 候选和“可能已解决”分类的先证者需要进一步评估/功能研究以确认致病性,从而为发现研究和药物洞察带来相应的好处。28% 的先证者未发现令人信服的变异,这代表了一个重要的新多样化发现队列,需要进一步分析,包括全基因组和长读方法。
ICG神经肌肉疾病(NMD)团队和成果依赖于过去十年间建立的小规模国际合作,建立信任和对当地人口、设施和观点的相互理解。此类合作受益于本地计算和数据及材料存储基础设施、小规模启动资金、促进临床和基因数据互操作性的举措以及基因匹配平台。该伙伴关系具有额外的双向利益潜力,包括加深对 VUS 的了解,这与所有家庭的 神经肌肉疾病(NMD)患者都息息相关,并且该伙伴关系是建立扩大检测能力、量身定制的护理指南和临床试验准备的基础。
总之,神经肌肉疾病基因检测证明了使用基于云的平台建立虚拟的国际协作伙伴关系并利用大数据来描述从许多中低收入家庭招募的多样化队列中存在的表型和致病变异是可行的。超过一半的测试参与者获得了基于研究的基因诊断,为患者提供了正确的 DNA 诊断的潜在益处,并证明了在临床试验中纳入更多不同人群的可行性。神经肌肉疾病基因检测认识到这项研究存在局限性。神经肌肉疾病基因检测没有试图收集流行病学数据,神经肌肉疾病基因检测的研究不允许得出关于神经肌肉疾病(NMD)或基因的发病率或流行率的结论。另一方面,这项研究是在“真实世界”的环境中进行的,反映了每个中低收入家庭中心的当前实践,并表明尽管存在局限性,但在这个以前未进行过基因测试的人群中,跨神经肌肉疾病(NMD)解决率可以达到 44-59%。这项工作表明,通过这种虚拟伙伴关系可以潜在地解决获得正确 DNA 诊断的地理不平等问题。这些对于所有合作伙伴和更广泛的研究界来说具有真正的双向价值。增加对遗传多样性的了解可以提高对变异的高效解释,从而提高低收入和高收入环境中患者的遗传诊断的正确性。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
