












【佳学基因检测】视网膜色素变性基因检测——助力准确诊断
视网膜色素变性的诊断为什么需要反复确诊?
视网膜色素变性是一种视网膜的遗传性疾病,其表现形式可以有很多种多样的特征,主要包括以下几点:
-
夜盲症(Night Blindness):最常见的症状之一,患者在低光条件下视力显著下降,特别是在夜晚或弱光环境下。
-
中心视力减退:患者在中央视野区域的视力逐渐减退,最终可能导致中心视觉丧失,这种情况称为中心性视网膜色素变性。
-
眼底骨膜变薄:眼底骨膜变薄是视网膜色素变性的早期体征之一,可以通过眼底检查来观察到。
-
视力波动:视网膜色素变性患者的视力可能会出现波动,即使在同一天内也可能有所不同。
-
眼底色素沉着:视网膜色素变性患者眼底部分区域可能会有色素沉积,这通常可以通过眼底检查来看到。
-
视野缺损:视网膜色素变性可能导致视野的逐渐缩小或存在不同程度的视野缺损。
-
色觉缺陷:一些患者可能在色觉方面有所损害,表现为对某些颜色的辨别困难或色彩感知异常。
-
眼底动脉变细:视网膜色素变性也可能伴随着眼底动脉变细的表现。
这些表现形式的具体情况可以因人而异,也受到具体遗传突变类型的影响。诊断和治疗视网膜色素变性通常需要眼科专家进行详细的眼底检查和遗传咨询
视网膜色素变性与多种眼科疾病如何在临床表征上相互重叠给诊断带来困难?
视网膜色素变性在临床表征上与多种其他眼科疾病有时会相互重叠,这可能给诊断带来一些困难。以下是一些可能导致混淆的情况:
视网膜色素变性与视网膜色素上皮炎(Retinal Pigment Epitheliopathy):这两者在眼底检查时可能表现出类似的色素沉着和视网膜改变,但前者是一种常见的眼底变性疾病,后者是一种比较由佳学基因检测进行检测与分析炎症性疾病。鉴别需要依赖于临床表现的详细分析和可能的遗传学检测。
视网膜色素变性与遗传性视网膜病变:某些遗传性视网膜病变,如遗传性黄斑变性等,也可能表现为视力减退、视野缺损和眼底色素沉着。区分这些疾病可能需要基因检测和详细的家族史调查。
视网膜色素变性与视网膜血管疾病:一些视网膜血管疾病如中心性视网膜静脉阻塞或视网膜动脉硬化也可能导致视力减退和眼底改变,这些症状有时与视网膜色素变性相似,需要通过血管造影和视网膜血流动力学研究来帮助鉴别。
视网膜色素变性与视网膜色素病变:某些局限性的视网膜色素病变如色素病变性视网膜脱离等也可能表现为视力减退和眼底色素沉着,与视网膜色素变性有时难以鉴别。
因此,准确诊断视网膜色素变性需要综合利用眼底检查、视野检查、色觉检查以及可能的遗传学和分子生物学检测。临床医生需要仔细分析患者的病史和临床表现,有时还需要长期的跟踪观察,以确定最终的诊断。
基因检测基因序列突变可以获得视网膜变性的确切证据
非综合征性视网膜色素变性
常染色体显性视网膜色素变性
眼科疾病的致病基因鉴定基因解码至少收录了24 种不同基因的突变与常染色体显性视网膜色素变性有关,一些基因的突变在视网膜色素变性病例所占的相关百分比相当高,其中包括RHO(26.5%)、RPRH2(5-9%)、PRPF31(8%)和RP1(3-5%)。而另一些所占的比例很低,但是它们仍然可能是受检者发病的真正原因。下表列出与常染色体显性视网膜色素变性相关的部分基因。
表1:与常染色体显性视网膜色素变性相关的基因
|
已鉴定基因 |
染色体定位 | 基因的功能 | 其他表型 |
| S*P2 | 2q37.1 | 它可以协调骨转换的某个方面 | 没有任何 |
| O*2W3 | 1q44 | 启动触发嗅觉的神经反应 | 没有任何 |
| H*1 | 10q22.1 | 有助于糖酵解和糖异生,能量通路 | 过度非球形红细胞溶血性贫血,隐性遗传性神经病(Russe) |
| B*ST1 | 11q12.3 | 提供制作 Bestrophin 的说明 | 隐性视网膜色素变性 |
| C*4 | 17q23.2 | 参与呼吸、钙化、酸碱平衡 | 没有任何 |
| C*X | 19q13.32 | 维持正常的视杆和视锥功能 | 显性莱伯先天性黑蒙 |
| TO**RS | 9p21.1 | 它具有与肿瘤抑制蛋白 P53 相互作用的能力 | 没有任何 |
| F**N2 | 17q25.3 | 充当肌动蛋白捆绑蛋白 | 没有任何 |
| G**A1B | 6p21.1 | 刺激鸟苷酸环化酶 1 和鸟苷酸环化酶 2 | 显性黄斑营养不良 |
| IM**H1 | 7q32.1 | 催化肌苷 5′-磷酸 (IMP) 转化为黄苷 5′-磷酸 | 显性莱伯先天性黑蒙 |
| N*L | 14q11.2 | 调节 RHO 和 PDE6B 基因表达的转录因子 | 常染色体隐性视网膜色素变性 |
| N**E3 | 15q23 | 视杆细胞的发育和视锥细胞的发育抑制 | 隐性视网膜色素变性 |
| S**A4A | 1q22 | 细胞表面受体 | 显性视锥细胞-视杆细胞营养不良症 |
| RHO | 3q22.1 | 出生后感光细胞存活所必需的 | 隐性视网膜色素变性 |
| P**H2 | 6p21.1 | 对椎间盘形态发生至关重要 | 具有 ROM1 的二基因形式 |
| R**65 | 1p31.2 | 在 11-顺式视网膜的产生和视觉色素再生中的作用 | 没有任何 |
| P**F8 | 17p13.3 | 作为介导剪接体蛋白和 snRNA 有序组装的支架 | 没有任何 |
| ROM1 | 11q12.3 | 对椎间盘形态发生至关重要 | PRPH2型双基因视网膜色素变性 |
| P**F31 | 19q13.42 | 参与前 mRNA 剪接 | 没有任何 |
| K**L7 | 7p15.3 | 介导 Lys-48' 连接的泛素化 | 没有任何 |
| R*1 | 8q12.1 | 感光细胞分化所需 | 隐性视网膜色素变性 |
| P**F4 | 9q32 | 参与前 mRNA 剪接 | 没有任何 |
| P**F3 | 1q21.2 | 参与前 mRNA 剪接 | 没有任何 |
| RDH12 | 14q24.1 | 在视锥细胞视色素再生过程中,由 11-顺式视黄醇形成 11-顺式视黄醛的关键酶 | 隐性莱伯先天性黑蒙症 |
| PRPF6 | 20q13.33 | 参与前 mRNA 剪接 | 没有任何 |
| RP9 | 7p14.3 | 与 PIM1 相关的 B 细胞增殖作用 | 没有任何 |
| SNRNP200 | 2q11.2 | 参与剪接体的组装、激活和拆卸 | 没有任何 |
PRPF31(前 mRNA 加工因子 31)
基因解码认为PRPF31 (61 kDa 的前 mRNA 剪接因子)突变在常染色体显性视网膜色素变性中起着至关重要的作用,诱发 1-8% 的常染色体显性视网膜色素变性病例
显然,PRPF31是诱发常染色体显性视网膜色素变性的三个前 mRNA 剪接因子之一,另外两个因子是PRPF3(占所有病例的 1%)和PRPF8 (占所有病例的 3%)。 PRPF31突变的独特特征之一是不完全渗透。遗传方式可能很复杂,无法确定无症状携带者是否影响了父母和孩子,因为家庭的遗传咨询受到阻碍。据报道,有症状的患者在青少年时期会出现夜盲症和视野丧失,并且通常在 30 多岁时被报告为失明。无症状和有症状患者的单倍型分析比较表明,两种类型都遗传了不同的野生型等位基因。高表达的野生型等位基因可以适应无功能的突变等位基因,但低表达的野生型等位基因无法达到所需的光感受器特异性PRPF31的活动阈值水平。
RP1(视网膜色素变性 1)
通过致病基因鉴定基因解码,RP1基因通过连锁检测在一个大型常染色体显性视网膜色素变性家族中被发现。RP1基因突变可诱发显性和隐性类型的视网膜色素变性。RP1 基因编码 240 kD 的视网膜光感受器特异性蛋白,其表达非常显著。携带 RP1 突变基因的患者的临床诊断显示视野直径缩小。一般来说,遗传性疾病被认为是由环境因素、等位基因异质性和遗传变异引起的,但对于 RP1 疾病,遗传变异被认为很重要,因为疾病的严重程度各不相同,患者可能具有相同的原发突变。
RHO(视紫红质)
眼科疾病的发病原因解析指出视觉传导通路的第一个成分是视紫红质,当光被视网膜的视杆细胞吸收时,视紫红质就被激活。常染色体显性视网膜色素变性有超过100个突变,约30–40%的病例是由于RHO基因突变引起的。常染色体隐性视网膜色素变性和常染色体显性先天性夜盲症也可由RHO基因突变引起。《眼科基因检测基因列表》收录了一例常染色体显性视网膜色素变性病例,该基因突变导致一种蛋白质没有第6和第7个跨膜,包括11-顺式视网膜结合位点。在携带显性视网膜色素变性的小鼠模型实验中,视网膜下注射含有 RNAi 抑制剂的重组非相关病毒载体后,视网膜功能得到改善。最近,在 P23H 转基因大鼠的视网膜下注射含有 Bip/Grp78 基因的重组非相关病毒载体后,视觉功能得到恢复。但这些先进的疗法需要以基因解码为基础的分子诊断作为治疗依据。由于人们对RHO基因突变引起的常染色体显性视网膜色素变性有了充分了解,治疗方法也在以同样的速度发展。
PRPH2(周围蛋白 2)
PRPH2 (Peripherin 2)基因曾被确认为RDS (视网膜变性慢变性)基因,含有3个外显子,编码346个氨基酸的蛋白质(39 kDa的整合膜糖蛋白)。该蛋白位于视锥和视杆感光细胞的外节盘,含有一个盘内结构域(D2)和四个跨膜结构域(称为M1-M4)。该蛋白与另一种蛋白(ROM1)结合形成同四聚体和异四聚体复合物,也能与其自身形成同寡聚结构。《视网膜色素变性基因检测案例集》收录了PRPH2突变诱发视网膜色素变性,基因检测数据分析指出约5-9%的常染色体显性视网膜色素变性病例由该基因引起。PRPH2基因和ROM1基因突变已被观察到可导致双基因视网膜色素变性。
常染色体隐性视网膜色素变性
对于常染色体隐性遗传性视网膜色素变性,《眼科疾病的致病基因鉴定基因解码》早就对至少40个基因的发病位置进行了北斗导航级高精度定位。其中大多数基因的突变容易被基因检测包所忽视。因为它们是由佳学基因检测进行检测与分析,导致 1% 的病例。但有一些基因如RP25、PDE6A、RPE65和PDE6B 的患病率较高,占所有病例的 2-5%。
表2:与常染色体隐性遗传性视网膜色素变性相关的基因
| 已鉴定基因 | 基因功能 | 染色体定位 | 隐性视网膜色素变性以外的表型 |
| ADIPOR1 | ADIPOQ 受体,维持正常的葡萄糖和脂肪稳态 | 1q32.1 | Bardet-Biedl 类 |
| P**GNT1 | 参与O-甘露糖基化 | 1p34.1 | 没有 |
| ZNF408 | 可能参与转录调控 | 11p11.2 | 显性家族性渗出性玻璃体视网膜病变 |
| NE**OD1 | 神经发生调节剂 D1,作为转录激活剂 | 2q31.3 | 没有 |
| IFT172 | 纤毛维持和形成所必需的。在刺猬信号传导中起间接作用 | 2p33.3 | 隐性 Bardet-Biedl 综合征 |
| I**140 | IFT 复合物 A 的组成部分,在纤毛细胞的正常发育和功能以及纤毛发生和纤毛的维持中起重要作用 | 16p13.3 | 隐性 Mainzer-Saldino 综合征、隐性 Leber 先天性黑蒙 |
| HGSNAT | 参与糖胺聚糖降解和聚糖结构降解 | 8p11.21 | 隐性粘多糖贮积症 |
| R**11 | 对类视黄酸具有氧化还原催化活性,参与短链醛的代谢 | 14q24.1 | 没有 |
| DHX38 | 胚胎发生、精子发生、细胞生长和分裂 | 16q22.2 | 早期发病的黄斑缺损 |
| KIZ | 需要稳定中心粒周围膨胀的中心粒周围物质 | 20p11.23 | 没有 |
| BEST1 | 阴离子通道 | 11q12.3 | 显性视网膜色素变性 |
| ABCA4 | 视网膜代谢 | 1p22.1 | 隐性黄斑营养不良 |
| AR**BP | 在核转运中的作用 | 16p13.3 | 没有 |
| C2orf71 | 可能在正常视力发育中发挥重要作用 | 2p23.2 | 没有 |
| C8***f37 | 未知 | 8q22.1 | 没有 |
| C**KL | 组织维护和发育 | 2q31.3 | 没有 |
| CLRN1 | 共轭 | 3q25.1 | 没有 |
| C**A1 | 光传导 | 4p12 | 没有 |
| CNGB1 | 光传导 | 16q13 | 没有 |
| CRB1 | 组织维护和发育 | 1q31.3 | 隐性莱伯先天性黑蒙症 |
| D**DS | 催化 | 1p36.11 | 没有 |
| D**38 | ATP 结合 RNA 解旋酶参与前 mRNA 剪接 | 16q22.2 | 没有 |
| EMC1 | 未知 | 1p36.13 | 没有 |
| EYS | 细胞外基质蛋白 | 6q12 | 未知 |
| F**161A | 未知 | 2p15 | 未知 |
| GPR125 | 孤儿受体 | 4p15.2 | 没有 |
| I**3B | 参与克雷布斯循环 | 20p13 | 未知 |
| IMPG2 | 视网膜细胞间基质的成分 | 3q12.3 | 未知 |
| K***1549 | 未知 | 7q34 | 没有 |
| KIZ | 未知 | 20p11.23 | 没有 |
| LRAT | 视网膜代谢 | 4q32.1 | 隐性莱伯先天性黑蒙症 |
| MAK | 在精子发生过程中发挥重要作用 | 6p24.2 | 没有 |
| M**TK | 跨膜蛋白 | 2季度13 | 没有 |
| MVK | 胆固醇生物合成途径中的调节位点 | 12q24.11 | 没有 |
| NEK2 | 参与控制着丝粒分离和双极纺锤体的形成 | 1q32.3 | 没有 |
| N**E3 | 转录因子 | 15q23 | 显性视网膜色素变性、隐性增强型 S 锥综合征 |
| NRL | 组织维护和发育 | 14q11.2 | 显性视网膜色素变性 |
| P**6A | 光传导 | 5q33.1 | 没有 |
| P**6B | 光传导 | 4p16.3 | 显性先天性静止性夜盲症 |
| PDE6G | 光传导 | 17q25.3 | 没有 |
| PRCD | 未知 | 17q25.1 | 未知 |
| PROM1 | 细胞结构 | 4p15.32 | 伴有黄斑变性的隐性视网膜色素变性 |
| RBP3 | 视网膜代谢 | 10q11.22 | 未知 |
| RGR | 视网膜代谢 | 10q23.1 | 显性脉络膜硬化 |
| RHO | 光传导 | 3q22.1 | 显性视网膜色素变性 |
| RLBP1 | 视网膜代谢 | 15q26.1 | 隐性博特尼亚营养不良症 |
| RP1 | 组织维护和发育 | 8q12.1 | 显性视网膜色素变性 |
| R**65 | 视网膜代谢 | 1p31.2 | 隐性莱伯先天性黑蒙症 |
| SAG | 光传导 | 2q37.1 | 隐性Oguchi病 |
| SLC7A14 | 3q26.2 | 没有 | |
| SPATA7 | 未知 | 14q31.3 | 没有 |
| TTC8 | 细胞结构 | 14q32.11 | 隐性 Bardet-Biedl 综合征 |
| TULP1 | 组织维护和发育 | 6p21.31 | 隐性莱伯先天性黑蒙症 |
| USH2A | 细胞结构 | 1q41 | 隐性 Usher 综合征 |
| ZNF513 | 表达因子 | 2p23.3 | 没有 |
Rp25
在进行致病基因鉴定基因解码时,即使是仅在少数人群中发现,也应当包括在受检者的基因检测里需。因为,即使没有在其他人身上出现,一旦在受检者身上出现,如果不检测,受检者得到的检测结果就是假阴性。在其他地理区域由佳学基因检测进行检测与分析突变可能是导致特定人群患常染色体隐性视网膜色素变性的常见原因,例如据报道, RP25基因座导致西班牙人群中 10% -20% 的常染色体隐性视网膜色素变性病例。
磷酸二酯酶6(PDE6A、PDE6B、PDE6G)
致病基因鉴定基因解码清晰地指出,一个 α、β 和两个 γ 亚基是PDE6复合物的重要组成部分;它编码的蛋白质在视杆光感受器视觉光转导中起着至关重要的作用。由于 G 蛋白光激活,复合物通过 cGMP 的水解过程维持细胞内 cGMP 水平。在视网膜色素变性病例中,诱导视杆光感受器死亡的过程被基因解码揭示,但人们认为 PDE6 活性低可能导致视杆-视锥细胞退化。为了使光感受器正常发挥作用,PDE6复合物的每个亚基都是必需的,事实上,对于常染色体隐性视网膜色素变性,PDE6A和PDE6B基因突变是诱发疾病的第二大常见原因。当使用瑞复得、希爱力或艾力达等药物抑制PDE6时,视力丧失是携带PDE6基因突变的杂合携带者的主要风险。近来有报道称PDE6G基因发生突变,导致常染色体隐性遗传性视网膜色素变性(早发性)。
Rpe65
RPE65(一种将全反式视黄酯水解为 11-顺式视黄醇的酶)对于视杆和视锥视觉所必需的视觉色素的重新形成至关重要,它在视网膜的色素层中表达。《如何避免眼科基因检测中出现假阴笥》, RPE65基因有大约 60 个突变,可导致隐性视网膜色素变性(2%)和莱伯先天性黑蒙(16%)。在三项临床前实验中,莱伯先天性黑蒙患者注射了包含人类RPE65 cDNA的腺相关病毒载体。视力得到了适度改善。这说明一个好的基因检测不仅可以帮助诊断,更可以为先进的基因疗法提供依据。
X连锁视网膜色素变性
X 连锁视网膜色素变性患者在疾病发展的早期阶段表现出严重的表型,约占所有视网膜色素变性病例的 10-15%。在一些病例中,还发现了耳聋、精子发育异常和呼吸道缺陷。目前,在 X 染色体上 六个已定位的基因位点 ( RP2、RP6、RP23、RP24、RP34 和 RPGR ) 中,已有两个基因位点 ( RP2 和 RP3 ) 被识别(表5)。 超过70%的X连锁视网膜色素变性患者存在RP3基因突变,RP3基因产物位于视杆光感受器的外部节段上。约10%–15%的患者因RP2基因突变而携带X连锁视网膜色素变性。
表 3:与 X 连锁视网膜色素变性相关的基因
|
已鉴定基因 |
基因功能 | 染色体定位 | 表型 |
| OFD1, RP23 | 参与纤毛的生物发生 | 2.2 | 没有 |
| RP2 | 参与β-微管蛋白折叠 | Xp11.23 | 没有 |
| R**R | 鞭毛内运输 | 11.4 | X连锁视锥细胞营养不良症、X连锁先天性静止性夜盲症 |
| RP6 | 未知 | Xp21.3-p21.2 | 没有 |
| R**4 | 未知 | Xq26-q27 | 没有 |
| RP34 | 组织发育和维持 | q28基因 | 没有 |
综合征性视网膜色素变性
视网膜色素变性通常是一种孤立问题;只影响一只眼睛,但在其他各种罕见情况下,视网膜色素变性也与其他疾病有关。例子可能包括 Usher 综合征、refsum 综合征、Bardet-Biedl 综合征。
Usher 综合征
Usher 综合征是一种常染色体隐性遗传病,与视网膜色素变性、听力障碍和有时的前庭功能障碍有关。失聪是该综合征最常见的疾病。不同人群的患病率为 1:12,000-1:30,000 人。10-30% 的常染色体隐性视网膜色素变性病例是由 Usher 综合征引起的。临床上,Usher 综合征分为三类:(1)Usher 类型 I(重度形式)、(2)Usher 类型 II(中度至重度形式)和(3)Usher 类型 III。佳学基因解码已收录了 12 个 Usher 综合征基因;Usher 类型 I 有 7 个基因,Usher 类型 II 有 3 个基因,Usher 类型 III 有 2 个基因(表 4)。
表 4:已确定 Usher 综合征基因
| 疾病 | 已鉴定基因 | 染色体定位 | 基因功能 |
| Usher 类型 I | MYO7A | 11q13.5 | 在外层感光盘更新中发挥重要作用,介导耳蜗毛细胞的机械转导 |
| CDH23 | 10q22.1 | 维持耳蜗毛细胞纤毛束的正常系统,介导耳蜗毛细胞的机械传导 | |
| PCDH15 | 10q21.1 | 维持正常的视网膜和耳蜗功能 | |
| U**1C | 11p15.1 | 正常听力所需,介导耳蜗毛细胞的机械转导 | |
| USH1G | 17q25.1 | 发育和维持耳蜗毛细胞束 | |
| CIB2 | 15q25.1 | 对感光细胞的正常维持和功能很重要 | |
| C**N1 | 3q25.1 | 在连接毛细胞和耳蜗神经节细胞的兴奋带中的作用 | |
| Usher II 型 | USH2A | 1q41 | 涉及听觉和视觉 |
| GPR98 | 5q14.3 | 受体可能在中枢神经系统发育中发挥关键作用 | |
| D**B31 | 9q32 | 对柯蒂氏器内毛细胞和外毛细胞的延长和维持至关重要 | |
| Usher类型 III | HARS | 5q31.3 | 负责组氨酰转移 RNA 的合成 |
| A**D12 | 2p11.21 | 可能调节内源性大麻素信号通路 |
Bardet-Biedl 综合征
Bardet-Biedl 综合征是一种隐性遗传疾病,症状包括肥胖(72%)、学习困难、手指/脚趾异常、肾脏疾病、视杆-视锥细胞营养不良(>90%)和肾脏异常。Bardet-Biedl 综合征影响 1:120,000 人,根据视网膜色素变性基因检测大数据分析,北大西洋人群、贝都因人(1:13500-1:16900)、中国人的患病率较高(1:13,000)。患有 Bardet-Biedl 综合征的儿童视力预后较差。夜盲症通常在 7 至 8 岁时显现。迄今为止,《眼科疾病的致病基因鉴定基因解码》已明确至少21 个基因的突变与 Bardet-Biedl 综合征相关。
表5:常染色体隐性 Bardet-Biedl 综合征的基因
| 已鉴定基因 | 染色体定位 | 基因功能 |
| ARL6 | 3q11.2 | 需要正常的视网膜功能和组织 |
| BBIP1 | 10q25.2 | 调节细胞质微管的恒定性和乙酰化 |
| BBS1 | 11q13 | BBSome 是纤毛发生所必需的,但不是中心粒卫星功能所必需的 |
| BBS2 | 16q12.2 | |
| BBS4 | 15q24.1 | 需要微管锚定在着丝粒上 |
| B*S5 | 2q31.1 | BBSome 是纤毛发生所必需的,但不是中心粒卫星功能所必需的 |
| B*S7 | 4q27 | |
| BBS9 | 7p14.3 | |
| B*S10 | 12q21.2 | 帮助蛋白质折叠 ATP 水解 |
| BBS12 | 4q27 | |
| C**290 | 12q21.32 | 触发 ATF4 介导的转录 |
| IFT27 | 22q12.3 | 具有 GTPase 活性 |
| INPP5E | 9q34.3 | 特别适用于脂质底物 |
| KCNJ13 | 2q37.1 | |
| LZTFL1 | 3p21.31 | 可能发挥肿瘤抑制作用 |
| MKKS | 20p12.2 | 帮助蛋白质折叠 ATP 水解 |
| MKS1 | 17q22 | 细胞分支形态要求 |
| NPHP1 | 2q13 | 与 BCAR1 结合控制上皮细胞极性 |
| SDCCAG8 | 1q43 | 建立细胞极性和上皮管腔形成 |
| TRIM32 | 9q33.1 | 可能介导 HIV-1 的生物活性 |
| TTC8 | 14q32.11 | 纤毛发生所必需的,但中心粒卫星功能所必需的 |
视网膜色素变性基因检测如何助务眼疾病的诊断
全外显子组测序是临床和症状遗传学的有效工具
DNA测序技术的进步已成为基础生物学和临床研究最有效的资源,并已应用于生物系统学、诊断学、生物技术、亲子鉴定和法医鉴定等各个领域。链终止测序和聚合酶链式反应的结合促成了许多技术的显著进步,例如人类基因组计划的完成,这为研究相关表型的基因修饰提供了足够的参考资料。最近,全基因组测序和全外显子组测序的新技术以低成本的每外显子组/基因组测序成本取代了传统方法。这些先进的下一代测序技术彻底改变了临床结构,改善了人类健康,尽管仍有许多问题需要解决,例如程序成本高、用于分析原始遗传学和序列数据的用户友好型软件以及与收集遗传数据有关的伦理问题。
全外显子组测序在人类遗传疾病中的作用
根据 OMIM及《人类疾病表征及其基因原因》的统计数据,基因检测数据库已收录超过 6000 种的单基因疾病,将近三分之二的疾病可以能过致病基因鉴定基因解码来明确。寻找表型变异和致病基因有助于了解现行疾病的致病机制。对于新发现变异的患者或小家族,仅凭发现的变异很难进行基因诊断。如果疾病非常罕见,则很难找到更多的患者。需要借助功能实验来验证最近报道的变异是否具有病理效应;如果突变基因在与疾病相关的已知途径中具有明确的功能,则可以进行生化确认实验。鉴定导致罕见单基因疾病的新基因对于了解导致疾病的生物学途径以及治疗管理非常重要。最近的研究强调,全外显子组测序是找出导致孟德尔疾病的偶然基因的有力技术;图。 2表明外显子组测序与过滤策略相结合有助于辨别出导致孟德尔遗传疾病的根本基因。
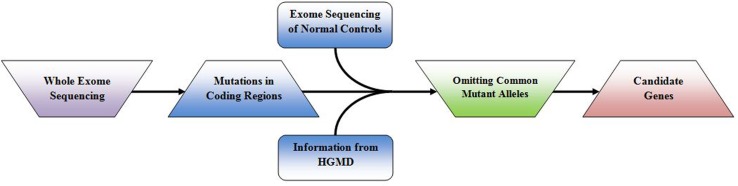
图 1:基因解码过程中的外显子组测序和致病基因筛选策略
异质性单基因表型
有许多遗传异质性疾病,如视网膜色素变性、智力障碍、遗传性听力障碍和自闭症谱系障碍。全外显子组测序已成功区分导致视网膜疾病的各种基因。表5展示了全外显子组测序在识别视网膜疾病新发基因中的作用。显明展示出基因解码基因检测优于基于数据库比对的基因检浊
表 5:全外显子组测序+基因解码有能力找出新的致病基因
| 疾病(视网膜疾病类型) | 染色体定位 | 基因鉴定 | 参考文献 |
| 隐性黄斑营养不良 | 1p13.3 | DRAM2 | El-Asrag et al. (2015) |
| 隐性全色盲 | 1q23.3 | ATF6 | Ansar et al. (2015), Kohl et al. (2015), Xu et al. (2015a) |
| 显性视网膜色素变性 | 1q44 | OR2W3 | Ma et al. (2015) |
| 隐性 Bardet-Biedl 综合征;隐性视网膜色素变性 | 2p33.3 | IFT172 | Bujakowska et al. (2015) |
| 隐性视网膜色素变性 | 2q31.3 | NEUROD1 | Wang et al. (2015) |
| 显性视网膜色素变性 | 2q37.1 | SPP2 | Liu et al. (2015) |
| 隐性眼耳综合征 | 4p16.1 | HMX1 | Gillespie et al. (2015) |
| 隐性小头畸形、生长障碍和视网膜病变 | 4q28.2 | PLK4 | Martin et al. (2014) |
| 隐性视网膜色素变性、非综合征性隐性粘多糖贮积症 | 8p11.21 | HGSNAT | Haer-Wigman et al. (2015) |
| 隐性视网膜营养不良伴虹膜缺损 | 9q21.12 | MIR204 | Conte et al. (2015) |
| 显性视网膜色素变性;隐性非球形红细胞溶血性贫血,隐性遗传性神经病 | 10q22.1 | HK1 | Sullivan et al. (2014), Wang et al. (2014) |
| 显性家族性渗出性玻璃体视网膜病变;伴有玻璃体改变的隐性视网膜色素变性 | 11p11.2 | ZNF408 | Avila-Fernandez et al. (2015), Collin et al. (2013) |
| 隐性视锥杆营养不良症;隐性乔伯特综合征 | 12q21.33 | POC1B | Beck et al. (2014), Durlu et al. (2014), Roosing et al. (2014) |
| 隐性视网膜色素变性 | 14q24.1 | RDH11 | Xie et al. (2014) |
| 隐性视锥细胞和视锥杆细胞营养不良症 | 14q24.3 | TTLL5 | Sergouniotis et al. (2014) |
| 隐性脉络膜视网膜病变和小头畸形 | 15q15.3 | TUBGCP4 | Scheidecker et al. (2015) |
| 隐性视网膜营养不良和小脑发育不良 | 18p11.31-p11.23 | LAMA1 | Aldinger et al. (2014) |
| 伴有脉络膜视网膜营养不良的隐性 Boucher-Neuhauser 综合征 | 19p13.2 | PNPLA6 | Kmoch et al. (2015), Synofzik et al. 2013, Topaloglu et al. (2014) |
| 隐性视网膜色素变性 | 20p11.23 | KIZ | El Shamieh et al. (2014) |
| 隐性 Usher 综合征 | 20q11.22 | CEP250 | Khateb et al. (2014) |
视网膜色素变性基因检测致力于不断改进和提高
由于基因解码技术的不断推进,人们希望引入新的策略用于临床实践的视网膜色素变性的分子诊断,并发现致病基因变异。但要实现这一愿望,需要满足特定条件:(1)应报告所有致病基因变异;(2)分子诊断技术应低成本、可靠、快速且广泛可用;(3)临床应能够理解疾病分子诊断提供的分子信息。目前已有可靠的技术可用,而且新技术正在兴起,这增加了报告个人新突变的机会。对于已知突变检测,目前基于阵列的诊断技术可用于多种视网膜疾病。下一代测序使研究人员能够识别致病变异并报告特定疾病的新基因。更新技术最近已被用于检测引起常染色体显性视网膜色素变性的基因和变异,以提升传统方法。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
