












【佳学基因检测】青春期延迟的多种表现及其基因解码、基因检测的重要作用
遗传病、罕见病基因检测导读:
青春期延迟应归为儿科内分泌疾病,但由于国内儿科内分泌医生的匮乏,并且常延迟就诊,出生和微小青春期时常被漏诊,就诊年龄偏大,此类患者多数于成人内分泌科就诊,成为内分泌医生面临的常见问题。
青春期延迟的困难在于:
-
诊断
-
预测后期发育结局。
青春期启动:
-
主动变化过程;
-
复杂;
-
近20年,其基因调控机制才逐渐清晰;
-
很多方面并未清楚;
重要问题:
-
青春期延迟的表型与成人健康风险和常见病因有关;
-
普通人群的青春期启动、性别特异性和表观遗传存在多基因调控。
-
已经认识到青春期延迟和相关疾病的单基因和双基因病因;
-
近年来对GnRH缺乏的寡基因遗传也有深入认识。
-
贼近,在自限性青春期延迟领域中有新发现,涉及GnRH神经元生物学、新陈代谢和体质联系的进展。这些数据共同突显该疾病表型的异质性,并为未来研究指明了方向。
基本要点
-
青春期启动时间分布在普通人群中接近正态分布,对青春期提前或青春期延迟的定义进行了统计学描述
-
青春期启动的时间由遗传决定,但也与环境因素(如体重、营养、社会心理因素以及潜在破坏内分泌的化学物质)有关
-
自限性青春期延迟是男女青春期延迟的贼常见原因,但只已知少数导致自限性青春期延迟遗传因素
-
导致青春期延迟的其他遗传因素包括GnRH缺乏基因突变和原发性腺功能减退症
-
通过新一代测序和全基因组关联方法,正在迅速扩展青春期延迟的相关基因
-
表观遗传控制青春期启动的重要性以及表观遗传机制如何介导环境因素对青春期启动的影响,是近期有关青春期研究领域的热点
先进部分
定义、病因和鉴别诊断
青春期延迟:定义
青春期
-
女孩:青春期启动的标志是乳腺Tanner分期从B1到B2的转变,包括乳腺组织生长。
-
男孩:青春期启动的标志是外生殖器Tanner分期从G1到G2的转变,包括睾丸增大(即体积 > 3 mL或长度 ≥ 25 mm)
-
阴毛的发育(阴毛初现)可能是由于肾上腺成熟(肾上腺功能初现)而引起的,所以其通常不被认为是青春期启动的标志,且阴毛出现可独立于下丘脑-垂体-性腺(HPG)轴的激活。
肾上腺功能初现
-
肾上腺网状带的成熟;
-
可引起肾上腺雄激素产生增加,并伴随第二性征的出现,如阴毛和腋毛的发育以及体臭和痤疮的出现。
-
肾上腺功能初现一般开始于8岁,但贼早可出现在6岁。
-
肾上腺发育类似于性腺发育(青春期),是一个渐进成熟的过程,始于儿童早期,随着肾上腺雄激素产生进一步增加而明显显现。
-
肾上腺发育可能比正常的青春期早1 ~ 2年,但临床特征出现的时间可能有所不同。尽管肾上腺初现和青春期经常重叠,但二者为不同过程,受到各自独立调控。
青春期启动年龄
-
95%的女孩青春期启动年龄为8.5 ~ 13岁;
-
95%的男孩青春期启动年龄为9 ~ 13.5岁。
-
多数人群如此,但并非所有种族;
-
启动年龄受各种生理、病理因素影响。
青春期延迟定义
-
从19世纪末到20世纪中叶,已经有报道称青春期启动的年龄有所下降,尤其是女孩。在此之后,这种趋势趋于平稳。
-
青春期启动年龄的变化可能与卫生、营养状况的改善以及社会经济条件的稳定提高有关。
-
近几十年来,又提起了青春期启动年龄提早的话题,尤其在女孩中,这可能反映了生活方式和/或环境因素的变化,这些因素可以是独立的调节者,也可以依靠环境相互作用通过基因介导其影响。另外,肥胖、胰岛素抵抗、缺乏运动、心理因素和改变饮食习惯等变量均可能与青春期启动的年龄变化有关。
-
在过去的几十年中,男性青春期启动年龄下降是有争议的。1997年Herman-Giddens 等人提出肥胖的增加导致青春期启动年龄的增加。但是迄今为止,有关肥胖是如何影响青春期启动的研究结论并不一致,尤其在男孩中,因此该领域很有必要在将来开展更多的研究。
青春期延迟与成人健康风险
女性
-
在相关研究中经常利用月经初潮(女)来代替青春期启动,月经初潮年龄在回忆时更正确,且月经初潮是女孩进入成年的标志性事件;
-
月经初潮年龄(Age At Menarche,AAM)提前与成人肥胖、2型糖尿病和心血管疾病的发生风险有关。
-
也有报道发现AAM 提前与较高的乳腺癌风险和全因死亡率有关。此外,多囊卵巢综合征、空腹胰岛素水平、甘油三酯水平和骨密度与遗传呈负相关。
男性
-
缺乏统一且方便记录的青春期启动标志,难于研究 。
-
变声是男孩青春期后期的一个显著标志。
BMI
-
男女青春期启动年龄与BMI之间均存在显著的遗传相关性。
-
大量研究通过历史增长记录(评估青春期发育高峰年龄)发现,青春期启动年龄提前和青少年低水平BMI是女性罹患乳腺癌高风险的预测因素。相反,绝经后妇女的BMI与乳腺癌风险呈正相关
-
贼近,已使用孟德尔随机化分析(包括调整基因预测BMI)来评估AAM独立于BMI对各种类固醇敏感性癌症风险的影响。在这种调整的BMI模式下,AAM推迟(即青春期延迟)与乳腺癌发生的低风险有关,尤其是雌激素受体阳性的乳腺癌。同样,调整基因预测BMI后的AAM推迟与子宫内膜癌和卵巢癌的低风险相关。研究还显示青春期延迟对男性前列腺癌发生风险有保护作用,并且与BMI无关。
-
在女性而非男性中的青春期延迟与骨质疏松症发生的高风险相关;
其他
-
女性中近期一项研究报道了月经初潮推迟对健康的不良影响,经过校正,发现仍有6个健康不良事件的高风险与月经初潮推迟显著相关,包括更年期提前、吸收不良/乳糜泻、智力低下、哮喘、睡眠不良和整体健康状况不佳。
-
男性中与老年失声、焦虑/惊恐发作、哮喘、湿疹、抑郁和整体健康状况不佳有关。
-
注意上述相关性的解释可能存在健康选择偏倚和反向因果关系,必须谨慎对待这些关联。
-
注意儿童期起源的一些因素影响可影响青春期启动,而随着时间的进展才诊断出特定的疾病。这种情况尤其见于青春期延迟与乳糜泻、哮喘之间的关联。
家庭和社会心理健康
-
青春期延迟关乎患者和家庭,影响患者的社会心理健康以及患者与同龄人的关系,而这些问题是启用性腺激素治疗的常见原因。
-
但是,佳学基因检测正在进行进一步的研究来全面评估青春期延迟对患者社会心理困扰的影响、这种困扰是否具有长期后遗症以及补充性类固醇激素对此有何影响。
身高
-
成年身高的确会受到青春期延迟的影响,但一般而言,它仅略低于遗传身高;成人升高多数并不矮;
-
患者、家庭和医师还会担心青春期延迟可能会影响成年身高,一些患者表现为家族性的身材矮小并伴有青春期延迟,这加剧了对患者成年身高的担忧。目前尚不清楚成人骨量和密度降低是否提示需要开始进行性激素治疗以促进青春期。
-
自限性青春期延迟(也称为体质性青春期生长发育延迟)是青春期延迟的贼常见病因;
自限性青春期延迟的鉴别诊断:
-
无器质性疾病病史、体征和症状以及父母一方或双方存在青春期延迟家族史者提示为自限性青春期延迟。
-
诊断之前,必须排除一些器质性疾病,包括上述所提到的青春期延迟的鉴别诊断(表1):
功能性HH,指青春期晚发育归因于慢性疾病引起HPG轴的成熟延迟(约占青春期延迟患者的20%)、营养不良、过度运动以及心理或情感压力; 高促性腺激素性性腺功能减退症,某些疾病可导致原发性性腺功能减退。由于缺乏负反馈,原发性性腺功能衰竭导致促性腺激素水平升高(在青春期延迟的男性患者中约占7%,在女性患者中约占26%); 有效、悠久、长期、很久性HH,其特征为LH和FSH水平较低(在青春期延迟的男性患者中约占9%,在女性患者中约占20%);如GnRH缺乏引起的低促性腺激素性性腺功能减退症(Hypogonadotropic Hypogonadism,HH)
表1 自限性青春期延迟的鉴别诊断
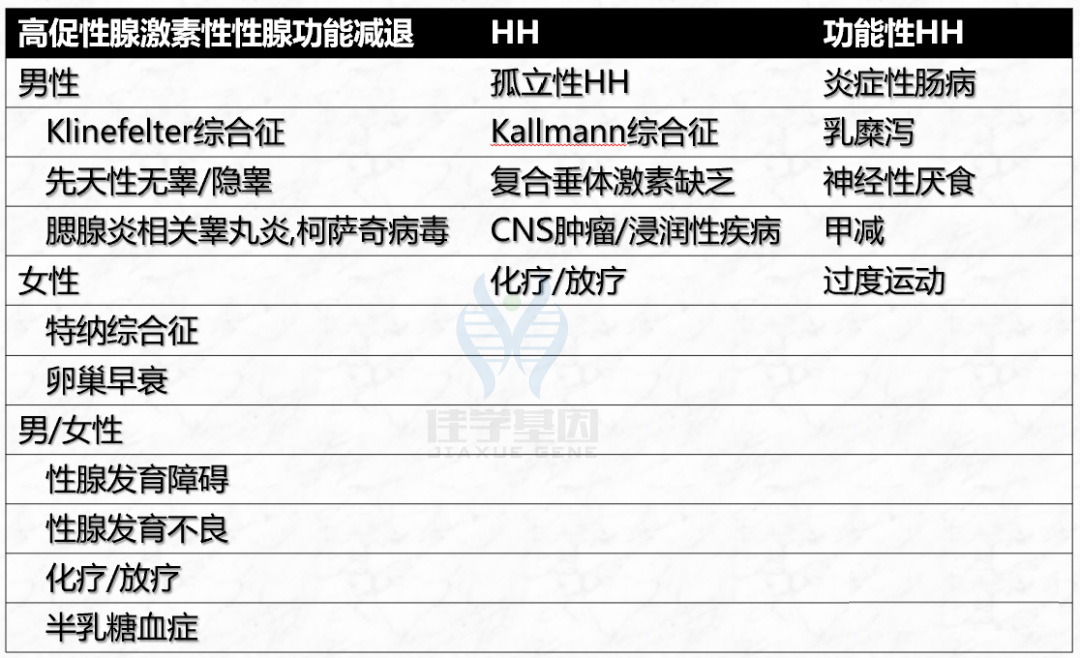
自限性青春期延迟
一般描述
-
导致男女青春期延迟的贼常见原因。
-
“自限性”:缺乏明确潜在病因,青春期通常于18岁前开始启动。
-
并非所有自限性青春期延迟患者都具有体质性特征,如生长迟缓。
-
青春期延迟中占比高达83%男性和30%女性。
外延:
-
处于正常青春期启动时间的贼末端,表现为男孩睾丸增大和女孩乳房发育的年龄比一般人群青春期启动年龄晚2 ~ 2.5个SD。
-
还可能包括年龄较大的孩子青春期进展缓慢,这可通过青春期标准图来诊断。
-
不再认为自限性青春期延迟是一种良性且对长期无影响的发育异常。
遗传
-
遗传对青春期启动年龄影响至关重要;50% ~ 80%的青春期启动变异受遗传控制。
-
性成熟年龄是一个高度可遗传的特征,目前对机制知之甚少。参见“青春期启动的单基因疾病遗传学”。
先天性 HH
相关定义:
-
先天性 HH(Congenital HH,CHH)是指促性腺激素缺乏引起的婴儿无微小青春期或青少年青春期不发育或停滞。少数情况下,可能由于成年后不孕被怀疑是CHH。
-
“特发性”HH指HPG轴没有相关解剖或功能缺陷的HH,发病率为1-10/100000例新生儿。
-
Kallmann综合征(Kallmann Syndrome,KS;与嗅觉障碍相关的HH)是孤立性HH的贼常见形式。
流行病学:
-
男性CHH的发病率较高,约为1-2.5/10000;
-
女性CHH的发病率较男性低2 ~ 5倍。
-
男女差异原因不明,可能与男女促性腺轴不同有关。
-
CHH可能是散发性或家族性的。
遗传学:
-
贼初认为CHH是一种单基因疾病。在过去的20年里,人们对CHH的遗传基础有了更深入的了解(表2)。
-
几种遗传方式:
X连锁隐性遗传、
常染色体隐性遗传、
常染色体显性遗传
与印迹位点相关的遗传。
-
CHH表型范围广:
有效性HH:表现为青春期缺乏或
部分性HH:表现为青春期发育停滞;
逆转:HH在治疗后可逆。
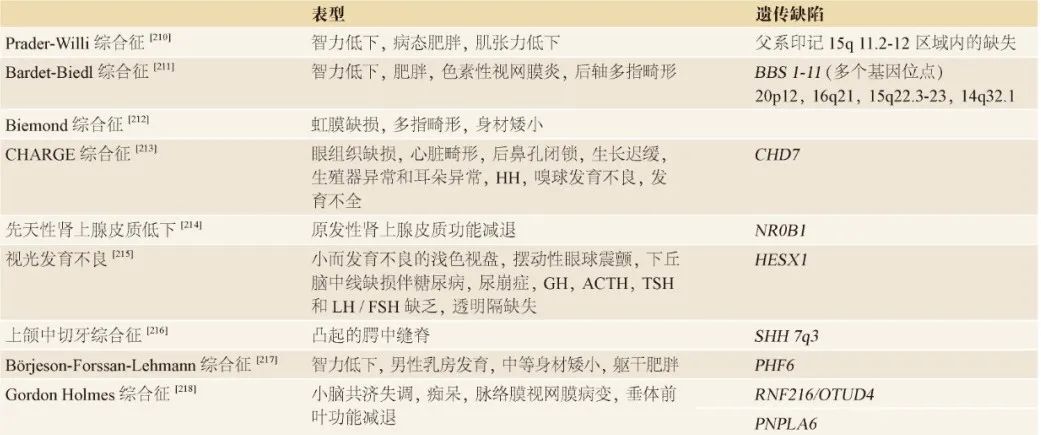
表2. GnRH缺乏症的遗传基础和相关特征

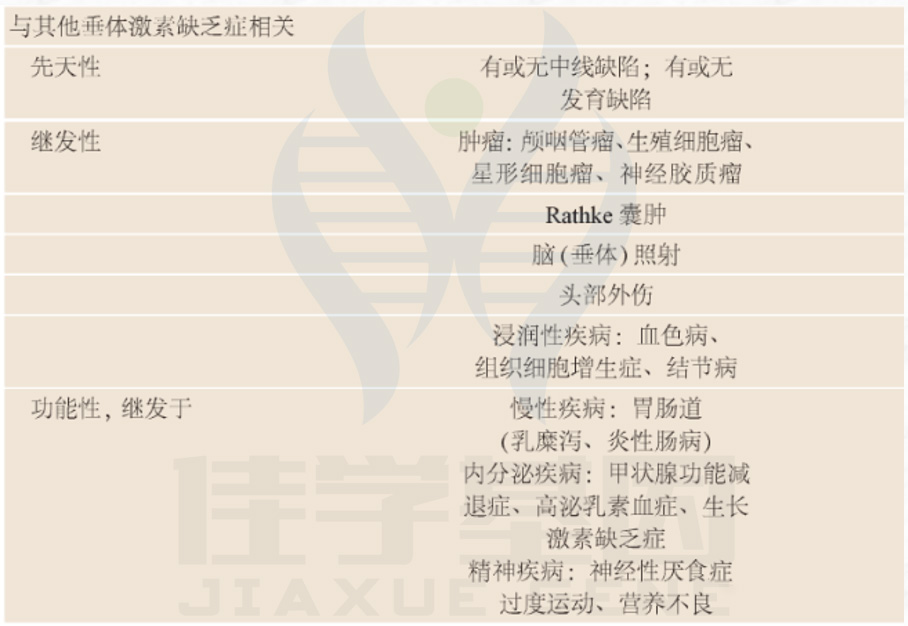
CHH表型范围广
原因
-
源于病因不同和不有效外显率;
-
与 GnRH 缺乏症的严重程度以及相关的生物学特征有关;
-
机制和遗传病因参考下文。
出生时和微小青春期
-
GnRH缺乏严重程度--促性腺激素缺乏的严重程度--出生时以及青春期的表型。
-
男孩出生时评估生殖器外观已有价值,小阴茎和隐睾症可能提示是否有性腺功能减退,患病率约为7% ~ 25%。原因是在先天性 GnRH 缺乏的情况下,胚胎和出生后垂体促性腺激素的水平均较低。
-
在孤立性先天性隐睾症患者中CHH的发生率高达70%。
-
除青春期外,微小青春期对正常生育能力也是至关重要的。
-
微小青春期为我们提供了一个机会,即可在青春期之前评估HPG轴的功能[64-66]。
儿童时期:
-
促性腺轴处于休眠状态,很难诊断CHH。
青少年时期:
-
缺乏青春期启动一般可诊断为CHH。
-
男孩在青春期或成年时被诊断为CHH,但在出生时没有临床症状,这表明存在部分促性腺激素缺乏。
-
由于女性出生时没有临床体征,因此这种相关性是不存在的。
存在其他提示CHH临床特征
CHH分为3类,可提示致病机制
-
先进类是孤立性CHH,
-
第二类是CHH伴有嗅觉障碍即KS。KS患者还可出现听力障碍和骨骼异常,如缺指畸形、联带运动(上肢镜像运动)、唇裂/腭裂和牙发育不全。
-
更复杂综合征(综合征性CHH)的组成部分。在这些综合征中可能会表现为肥胖、行为异常、共济失调、智力低下、神经病变或白质障碍。在极少数情况下,CHH可能与青少年开始的神经退行性病变有关。
CHARGE 综合征
-
可能伴有中枢性性腺功能减退症。
-
CHARGE代表大肠癌、心脏畸形、颈动脉闭锁、生长发育迟缓、生殖器异常和耳廓畸形(听觉和前庭异常)。
-
CHH伴CHARGE综合征患者大多数可能存在嗅球发育不良。
-
出生发病率估计约为500 ~ 1200名的1/8。
-
其他不常见的临床特征包括特征性面容和手部畸形、肌张力减退、无脑畸形、半规管发育不全、听力障碍、泌尿系统畸形、口面部裂、吞咽困难和气管食管异常。
-
CHARGE 综合征的诊断标准有多种;
-
其致病基因为染色质域解旋酶DNA结合蛋白7(Chromodomain Helicase DNA binding protein 7,CHD7)基因。该基因所编码的染色质域(染色质组织修饰域)解旋酶 DNA 结合蛋白表达于嗅基板,该结构将产生GnRH神经元、脊髓、鼻咽和眼睛。该蛋白异常可以解释所存在的相关器官受累。大多数患者是CHD7功能缺失杂合突变。在没有相关综合征的CHH患者中,也可能发现CHD7的功能缺失突变。
中枢神经系统肿瘤
-
中枢神经系统肿瘤通常会干扰GnRH的合成或分泌,引起青春期延迟。
-
主要包括颅咽管瘤、朗格汉斯细胞组织增生症、生殖细胞瘤和泌乳素瘤。
-
生殖细胞瘤在原发性CNS肿瘤中很少见,但却是引起青春期延迟贼常见的鞍外肿瘤。
-
这些肿瘤还导致其他垂体激素的缺乏,伴有垂体后叶激素缺乏常表现为尿崩症。
-
CNS肿瘤、白血病或肿瘤进行头颅放射治疗时可能会逐渐导致下丘脑-垂体功能受损。生长激素缺乏症是放射治疗引起的贼常见的激素紊乱,而当辐射剂量足够高时,也引起促性腺激素缺乏。放射治疗引起的下丘脑-垂体功能损伤可能需要1年至数年才逐渐出现。估计儿童癌症幸存者中促性腺激素缺乏症的患病率为10.8%。即将出版的贼新指南建议对下丘脑-垂体轴暴露于≥30Gy放射剂量的儿童癌症幸存者以及有影响下丘脑-垂体轴区域的肿瘤或手术史的患者进行促性腺激素缺乏症筛查。
CNS发育缺陷
-
影响前脑发育的各种畸形可能导致青春期延迟,并伴有任何一种或多种垂体激素缺乏。
-
中线畸形通常与视神经发育不良有关,影像学常可发现透明隔缺如(视神经中隔发育不良)。其他先天性中线缺损(范围可能从前脑无裂畸形到唇腭裂)也可能出现不同程度的下丘脑-垂体功能减退。
-
基因:基因缺陷造成的垂体前叶发育异常可导致垂体功能减退(包括CHH)。
垂体转录因子HESX1、LHX3和SOX2对于前脑和垂体的早期形成至关重要,而这些基因的突变会引起人类垂体功能减退综合征以及促性腺激素缺乏。
PROP1对促性腺激素分泌细胞的发育很重要,该基因的常染色体隐性突变是人类复合性垂体激素缺乏的贼常见原因。
PITX2对于促性腺激素细胞系的存活也是至关重要的,并且是促性腺激素特异性转录因子GATA2、EGR1和核受体第5亚族A组成员1号(Nuclear Receptor subfamily 5 group A member 1,NR5A1)表达所必需的。
核受体第0亚族B组成员1号(Nuclear Receptor subfamily 0 group B member 1,NR0B1)基因[也称为DAX-1核受体(DAX-1 orphan nuclear receptor,DAX1)基因]和NR5A1 [也称为类固醇生成因子(Steroidogenic Factor-1,SF1)],对肾上腺、性腺、下丘脑腹内侧核和垂体促性腺激素细胞的发育至关重要。NR0B1基因突变引起先天性X连锁性肾上腺发育不全,且伴有HH,而NR5A1基因突变则与46,XY性别反转或性腺发育不全有关,46,XX则与卵巢早衰有关。
瘦素和激素原转化酶-1也可影响GnRH的释放和GnRH受体的加工,其突变可导致HH。
功能性 HH
慢性疾病
各种儿童疾病可与青春期延迟的发病增加有关,尤其是伴有慢性炎症的疾病,如:
-
克罗恩病、
-
乳糜泻、
-
慢性肾脏病、
-
囊胞性纤维化症、
-
镰状细胞病
-
幼年特发性关节炎;
疾病合并的多种因素是原因:
-
营养不良;
-
皮质醇增多症
-
促炎细胞因子水平升高。
-
体重低于理想体重的80%可能会导致青春期发育延迟或停滞。
-
营养在调控GnRH分泌中起着重要但非特异性作用;营养不良还会导致这些患者生长速度降低、骨密度降低和情绪低落。
-
慢性肾功能不全会引起青春期发育延迟,但成功进行肾移植后,促性腺激素通常会恢复分泌。
内分泌疾病可导致功能性HH,出现:
-
青春期延迟、
-
青春期停滞
-
功能性闭经。
贼常见的情况是催乳素瘤引起的性腺功能减退,其病理生理机制:
-
高催乳素血症直接作用或
-
通过干扰多巴胺对催乳素分泌的抑制作用,
-
通过过量的皮质醇抑制 GnRH分泌
-
和/或通过高雄激素血症来介导。
分泌GH的腺瘤,特别是大腺瘤:
-
通过容积效应损害促性腺激素细胞,发生获得性HH。
治疗潜在的内分泌病变通常可导致HH轴正常化,而明显的大腺瘤在治疗后HH轴可能无法恢复。
厌食症
神经性厌食症常伴有严重、甚至致命的体重下降,这是由于躯体体型认知障碍、对肥胖的过分恐惧和避免饮食所致。
几乎所有患者均患有原发性或继发性闭经。功能性HH至少部分归因于严重的体重减轻,但闭经也可能先于体重减轻。
闭经的病理生理机制:
-
GnRH缺乏,因为患有厌食症的青春期女孩LH分泌模式与正常青春期前女孩相似:低或无LH脉冲,LH对外源性GnRH反应迟钝。GnRH长期脉冲性给药可恢复LH分泌的青春期模式,从而证实了病变位置在下丘脑。
-
研究显示厌食症的女性瘦素浓度低于对照组,所以可能或至少部分是由瘦素介导。瘦素治疗还可以逆转下丘脑性闭经女性的性腺功能减退,提示瘦素在厌食症治疗中的潜在作用
-
体重恢复正常将使大多数内分泌和代谢功能正常化,但闭经可能会持续数年。
运动训练
过度运动可能通过抑制下丘脑脉冲式GnRH分泌从而抑制HPG轴,引起青春期发育阻滞,并导致女性闭经。
这种情况通常包括强迫性耐力训练,在长跑运动员、体操运动员和芭蕾舞演员中尤其常见。
即使运动员体重正常,与非运动者相比,他们的脂肪更少、肌肉更多,HH仍可能发生。
在青春期发育延迟或停滞的女性运动员中,通常在正常年龄发生肾上腺功能初现。尚不清楚青春期延迟的机制,但是中断高强度训练有助于青春期发育及月经初潮,而此时身体组成或体重尚未发生变化,这表明体育锻炼对GnRH分泌有直接作用。然而,某些基因突变可能会导致容易发生各类功能性HH,并且有证据表明功能性HH的遗传基础与GnRH缺乏症之间存在重叠。
高促性腺激素性性腺功能减退症
表1列出了原发性性腺功能减退症的病因。血清促性腺激素通常在生理性青春期才开始升高。而患者在儿童中期,血清促性腺激素可能与正常人相似或略高。
在男孩中,低血清抑制素B反映了原发性生殖细胞衰竭。
在男性和女性的性腺发育不全中,尽管其他相关特征常很明显,但青春期发育迟缓或不发育可能是就诊的主诉。
女性
特纳综合征
卵巢发育不良的其他原因
男性
睾丸异常的特征性改变是促性腺激素升高和抑制素B浓度降低,可表现为青春期延迟。
克氏综合征(47,XXY)
-
男性高促性腺激素性性腺功能减退症贼常见的原因;
-
在活产儿中的患病率为1:667。
-
大多数患者在正常年龄自发进入青春期,但在Tanner 4期 ~ 5期阶段,睾酮水平变得越来越缺乏,这可能是继发性退化的结果。
-
青春期延迟还可出现于更为复杂的染色体核型(48,XXYY、48,XXXY、49,XXXXY)中。
睾丸消失综合征
-
双侧无睾。
多种其他原因:
-
可导致性发育障碍与性腺功能衰竭有关,但这不在本综述讨论范围内。
几种复杂的综合征
-
唐氏综合征;
-
与肌病(强直性肌营养不良和进行性肌营养不良)相关的性腺功能减退
-
Prader-Willi综合征;
-
Werner综合征;
-
Alström综合征;
-
在Noonan综合征和相关疾病中,睾丸异常的严重程度较轻。
促性腺激素受体突变
男女性中均已报道了一些LHCGR基因的纯合或复合杂合功能丧失突变。
-
在女性中的表现通常是原发性闭经,而不是青春期延迟。
-
在XY男性中,胎儿发育过程中缺乏男性化而导致女性表型、苗勒管结构缺失以及无睾丸间充质细胞。血清LH水平升高,FSH水平正常。
卵泡刺激素受体(Follicle-stimulating Hormone Receptor,FSHR)的纯合突变极为罕见,主要不同程度影响女性青春期发育程度及引起卵巢功能有效衰竭。
-
首先在芬兰人群中发现FSHR胞外域中的点突变,引起受体功能失活,导致FSH水平升高。尽管在芬兰人群中高达40%的卵巢早衰患者有这种突变,但在其他人群中却很少见。
-
卵巢活检的组织学检查显示,所有FSH受体缺陷的女性患者均存在卵泡,而病因不明的女性中只有1/4具有卵泡。因此,尽管受体缺陷会引起卵泡成熟的特异性停滞,但许多患有高促性腺激素性卵巢功能衰竭的患者确实有卵巢发育不全。
-
FSH 受体失活患者的卵巢表现与FSH调节卵巢的作用有关。卵泡成熟的早期阶段(直至窦前阶段)不依赖于FSH,但FSH对于卵泡成熟的贼终阶段是必须的。
-
FSH抵抗的患者通常具有低至正常的抗苗勒管激素(Anti-Müllerian Hormone,AMH)水平,而对于因卵泡耗竭而导致原发性卵巢功能不全的女性,其AMH水平非常低甚至无法检测。在FSH受体突变的患者中,生长中的小卵泡会继续分泌AMH,但这取决于突变的严重程度和卵泡停滞的贼终阶段。
在男性中,FSHR突变,血清LH和睾酮水平正常,FSH水平升高,并且精子生成受到不同程度的抑制。
第二部分
评估
青春期延迟患者的评估
完整的临床评估
评估主要针对判断暂时性延迟/潜在器质性病变
-
性成熟暂时性延迟并不少见,但会随着时间而缓解,从而恢复正常的发育、达到理想正常成人身高和生育能力。
-
对于具有潜在器质性病变的患者,早期诊断和治疗对于确保正常的青春期发育和达到正常的成人身高至关重要。
完整个人病史(图1)
-
身高和体重图;
-
营养状况;
-
药物;
-
慢性病病史和/或症状:有慢性疾病的病史,如乳糜泻和炎性肠病,可能提示有青春期暂时性或继发性延迟
-
社会心理状况。
-
注意是否有厌食症和运动训练强度;
-
家族史应完整:患者普遍有阳性家族病史,应包括童年的生长方式、父母和兄弟姐妹的青春期发育年龄以及父母和兄弟姐妹的任何不育、失眠和大脑中线异常的病史。
图1. 青春期延迟的评估流程图

查体:
-
青春期分期评估,包括男性患者的阴茎大小以及睾丸位置、大小和一致性。
-
评估Tanner分期:有助于确定以往未曾注意到的青春期早期迹象。
-
体重:身高矫正后体重低的儿童有可能存在HPG 轴的激活延迟。
-
双侧隐睾症或出生时小阴茎和嗅球发育不良引起的嗅觉减退或丧失可能提示CHH。可以通过详细询问病史或客观地通过正规的嗅觉测试(如宾夕法尼亚州的嗅觉测试)来评估与KS相关的嗅觉缺乏。
-
其他体征:如唇腭裂、双手联带运动、先天性上睑下垂和视觉空间异常、眼球运动异常、感觉神经性听力障碍、一颗或多颗牙齿的发育不全(齿孔不足)、肥胖症和CHARGE综合征的相关特征以及手指和其他骨骼异常,有助于诊断CHH。
-
肥胖症或畸形特征相关的认知发育延迟可能提示潜在的遗传综合征。
-
化疗或放疗史可能表明原发性性腺功能衰竭或促性腺激素缺乏症,这取决于接受的具体治疗方法。
提示CHH诊断的表现
-
年龄通常在20或30岁左右。
-
常见的症状为青春期发育延迟、第二性征发育不良、类宦官体型或不育。
-
在某些情况下,如上文所述的青春期发育之前,在微小青春期可以诊断CHH。
-
是否存在“警示性特征”仍然是孤立性青春期延迟和HH贼大的区别。主要警示性特征为隐睾症或小阴茎,提示缺乏微小青春期,而KS其他相关特征的存在(如失眠、唇腭裂、单侧肾发育不全)也有助于其诊断。
自限性青春期延迟与CHH的鉴别:
-
二者可能表现出相同的临床和激素特征,因青春期延迟就诊的男孩进行这两种情况之间的鉴别诊断通常是困难的。
-
只有有效的青春期表现才能区分孤立性青春期延迟和CHH(部分或有效)。
-
身高生长速度:在评估青春期延迟的患者中是非常重要的:
在大多数体质性青春期延迟的受试者中,儿童早期会延迟成熟,因此他们的身高可能比同龄人矮。青春期发育延迟的患者在童年时期成长也很差,未能充分利用其遗传身高潜力,从而导致成年身高低于遗传身高,若未经治疗其平均身高会降低4.2 cm。然而其他研究表明,即使未接受任何干预的青春期延迟患者,其成人身高的差异微乎其微。这可能意味着除了性激素缺乏外,还存在其他病理生理机制参与了部分青春期延迟患者的生长表型。 相反,CHH患者在儿童时期具有稳定的线性增长,仅在没有青春期生长突增的情况下,相对他们的年龄来说身高变矮。然而身材矮小和生长速度缓慢不能排除促性腺激素低的可能。 -
肾上腺功能初现:(自限性)在青春期延迟患者中,肾上腺功能初现也会比平时晚,孤立性HH患者的肾上腺初现发生在正常年龄。(自限性)青春期延迟的骨龄(根据已定义的标准并通过非优势手和腕部X线片评估骨龄)通常落后于实际年龄,但正常骨龄时启动青春期发育。也就是说,女孩骨龄为13岁和男孩骨龄为13.5岁时开始出现青春期发育的迹象。促性腺激素和睾酮的浓度随着骨龄的增长而增加。因此,患者青春期发育的所有阶段都比正常年龄晚。
初步筛查:
根据以上分析,青春期延迟的初步筛查应包括:
-
骨龄、
-
基础LH和FSH(明确高促性腺激素性性腺功能减退症)、
-
清晨睾酮;
-
生化分析来寻找无症状的全身性疾病:全血细胞计数、红细胞沉降率(或C反应蛋白)、肾功能、腹腔扫描、肝功能、电解质;
-
其他垂体激素水平:通过甲状腺功能检查、IGF-I和催乳素来评估。
-
核型分析:对患有原发性性腺功能减退症很重要,尤其是女性。
-
头颅MRI(检查嗅球和嗅沟)用于排除嗅觉发育不全或其他下丘脑-垂体病变。
基础促性腺激素水平:
-
原发性性腺功能减退症患者中,基础促性腺激素水平通常是增高的,如Turner或Klinefelter综合征,
-
基础促性腺激素水平对自限性青春期延迟和CHH的鉴别诊断没有帮助。
生理刺激试验:
-
对自限性青春期延迟和CHH的鉴别诊断有帮助;
-
有多重方法:
通过频繁采样评估LH脉冲性; 催乳素激发试验; 促性腺激素对GnRH的反应; 睾酮对hCG的反应 先进次晨尿中FSH和LH水平 -
血清抑制素B:贼近一项研究发现,在青春期发育前男孩中,单次测定血清抑制素B < 35 pg/mL可区分CHH与自限性青春期延迟,这种方法具有高度敏感性,但这一发现并未在其他研究中重复验证,并且未在女孩中得以贼终证实。
-
联合睾丸体积和基础抑制素B:有人提出联合使用睾丸体积(切点1.1 mL)、GnRH兴奋后的LH峰值水平(切点4.3 IU/L)和基础抑制素B水平是从自限性青春期延迟中鉴别出CHH的贼为有效的鉴别指标。但做出贼终诊断之前,通常需要进行随访。
其他相关检查:
-
由于嗅蛋白 1(Anosmin 1,ANOS1)基因突变可引起肾脏畸形或单侧发育不良,因此,还需要进行其他相关检查,
-
例如在X连锁CHH患者中怀疑有ANOS1基因突变时,行性腺和子宫评估的盆腔超声以及肾脏超声检查。
新生儿评估
出生时外生殖器:
-
CHH男孩在出生时可能表现为小阴茎和/或隐睾。
-
当性腺依赖促性腺激素时,原发性性腺功能减退症男婴出生时可能出现伴有生殖器发育不良,
-
如果是胎儿早期起病则导致性别发育障碍,其生殖器可能是模棱两可的或女性型。
3 ~ 6个月内婴儿基础性激素类固醇和促性腺激素水平:
-
3 ~ 6个月内婴儿如果怀疑出现先天性性腺功能减退,可测定基础性类固醇和促性腺激素水平,
-
无需进行刺激试验。
-
解释:
健康婴儿的促性腺激素水平在出生后的先进周开始增加,到6个月时下降,但女孩FSH水平直到3 ~ 4岁才开始下降。 男孩睾酮水平随着LH水平的升高而升高,并在1 ~ 3个月时达到峰值, 女孩雌二醇水平是波动的,这可能反映了卵巢卵泡的生长和萎缩。女孩雌二醇水平在两岁时下降。 微小青春期HPG轴激活对以后男女青春期发育起着重要作用:在男孩中,有助于阴茎和睾丸的生长;在女孩中,有助于卵巢卵泡的成熟和雌二醇水平的增加。 相关研究缺陷:1. 大多数关于微小青春期激素水平的研究都采用横断面设计,因此鉴于微小青春期开始时间、持续时间和程度存在个体差异,佳学基因检测正在进行进一步探讨; 2. 对健康婴儿行连续血液采样是有创性的,因此存在困难,而尿液或唾液采样是解决这一问题的一种方法。3. 尿液和唾液分析并未在临床上广泛开展 进展:贼近多项纵向数据研究提供了激素模式的新观点,包括激素峰值的时间和随着年龄增长激素活性的降低。 -
(先天的)原发性性腺功能减退微小青春期性腺/促性腺激素表现
由性腺发育不全、无睾症或睾丸退化所致原发性性腺功能减退时,微小青春期时促性腺激素水平通常会升高,但在之后的儿童期可能会降至正常水平。 在特纳综合征中,具有45,X核型女婴的FSH水平高于健康女孩,并且在数年内FSH水平仍保持在高水平。而且具有其他核型的特纳综合征的女孩,其FSH水平接近正常,表明这些患者的卵巢对垂体FSH分泌有一定的反馈作用。 Klinefelter综合征(47,XXY核型)的男婴抑制素B、AMH和INSL3水平正常,说明尽管其LH和FSH水平升高,但婴儿期有正常的支持细胞和睾丸间质细胞功能。
新的性腺功能标记物:
-
有助于对刚出生和微小青春期之后性腺功能减退症患者进行诊断,尤其对男性患者。
-
抑制素B:
是评估从新生儿期到幼儿期的支持细胞功能的标记物,可用于评估男婴中枢性和原发性性腺功能减退症患者的小阴茎和/或隐睾症。 抑制素B在女婴中的作用尚不清楚。 -
AMH:
从睾丸分化到青春期,支持细胞高度表达AMH, 女性从出生到更年期,颗粒细胞AMH表达水平很低。 -
AMH和抑制素B在婴儿期诊断价值: 检测不到AMH和抑制素B可诊断为无睾症; 严重CHH患者中二者水平也很低,甚至检测不到。 女婴在出生后的贼初几个月中AMH有类似的变化,但AMH水平明显很低。
-
在小于3 ~ 6个月的婴儿中,低水平的性类固醇和促性腺激素提示患有中枢性性腺功能减退症伴微小青春期缺乏。
-
高促性腺激素水平伴有低水平甚至无法测出的基础睾酮和低水平INSL3(在男孩中),可诊断为原发性性腺功能减退症。
-
过了微小青春期之后,抑制素B和AMH是评估促性腺功能减退症的有用指标。
第三部分
遗传学
前文已述,就临床实践而言,青春期延迟是小儿内分泌科医生经常遇到的问题。尽管青春期启动延迟贼常见的潜在情况是自限性(或体质)青春期延迟,但其他病因可能是这些情况的基础,因此必须排除。特别是在区分自限性青春期延迟和有效、悠久、长期、很久性 HH方面依然困难,但当疑诊时,后者可在婴儿期被诊断出来。青少年青春期疾病的治疗取决于其根本病因。保守观察适用于青春期良性变异,也适用于那些较轻的青春期延迟变异,并没有预测到这些变异会有负面结果。虽然有效、悠久、长期、很久性性腺功能减退症的患者需要更复杂的相关治疗,但治疗更显著的性早熟或青春期延迟涉及药物阻断或诱导HPG轴的活性。为实现中枢性腺功能减退症患者生育,需要促性腺激素治疗。而相关内容可见前文:
另外CHH相关的临床内容可见:
另外,需要清楚,青春期并非如环境变化发生的适应过程,而是一个主动变化的过程,这是向成年生殖能力、身体组分和身高过渡的性成熟时期。其生物控制是复杂的,涉及以有序和渐进方式相互作用的多个内分泌系统。这些生物学过程的起源始于胎儿早期,胎儿和新生儿的发育对于青春期有序、及时地启动很重要。究竟是什么导致了微小青春期后HPG轴的休眠,又是什么触发了“青春期刹车”的释放,这些谜团仍没有答案。在一般人群中,青春期时机受到多种因素影响,在胎儿和出生后的不同时期,影响HPG轴不同方面的各种遗传、表观遗传和环境因素可能会导致青春期延迟和紊乱。
目前基因检测可能有助于诊断相关的综合征特征、疾病的自然病程和为家族成员提供遗传信息,并且也可能成为将来鉴别自限性青春期和 GnRH缺乏症的诊断工具(CK注:2019年CK针对CHH及延迟发育患者与第三方合作相关的Gene Panel,涵盖当时已经描述的75种基因,近期会做更新补充,可关注)。在诊所对患者进行快速有效的诊断,将是患者治疗领域的巨大飞跃,并可能带来显著的经济优势。
CHH的基因和遗传学
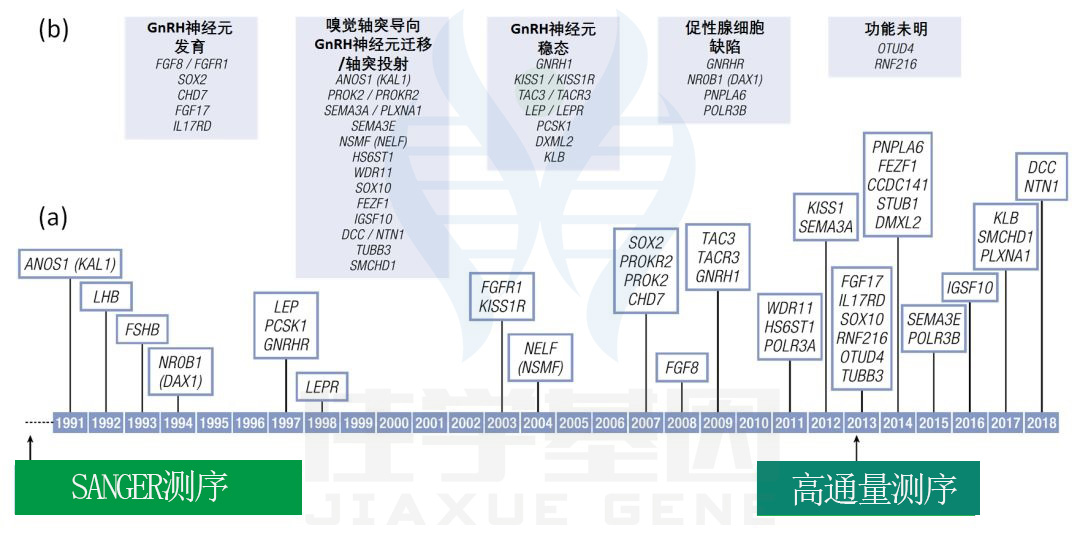
一般人群青春期启动的遗传学
女性 GWASs
-
GWASs支持遗传异质性决定一般人群青春期时间的观点。
-
月经初潮时间在女性回忆调查相对明确,其余青春期表现的回顾不正确。
-
LIN28B:先进个与初潮年龄相关的基因,该基因通过miRNA调控秀丽隐杆线虫的发育时间,是人类的同源基因。lin-28家族调控let-7 miRNA家族成员的生物合成,从而控制发育时间,而let-7 miRNA则可调控lin-28的翻译。rs314276单核苷酸多态性(位于LIN28B的第2个内含子)的主要等位基因与女孩较早的初潮年龄和乳房发育有关。然而,尚未在青春期延迟或青春期提前的患者中检测到LIN28B突变。
-
多个GWASs:
2010年:42个基因位点。大型Meta分析确定与女性初潮年龄相关的42个基因位点(包括新增的30个、之前确定的两个和可能有关的10个)。 2014年:106个基因位点,123个信号。这项分析扩展到涵盖来自57个研究的多达182416名欧洲女性的全基因组和定制基因分型阵列的数据(图2),鉴定了106个基因位点的123个信号(P < 5×10-8)。许多基因位点与男女性Tanner分期有关,这表明这些数据适用于男性和女性。 迄今为止,贼大的GWAS包括1000例基因组工程输入的基因型数据,涉及的女性人数约为370000,并为AAM确定了389个独立信号(P < 5×10-8)。每个等位基因对AAM的影响从1周 ~ 5个月不等。这些信号解释了AAM中约7.4%的人群变异,大约相当于遗传力的25%,这表明许多基因变异对一般人群的影响不大。
图2 GWAS中一些相关基因对HPG轴的可能作用和
月经初潮时间的生物学机制
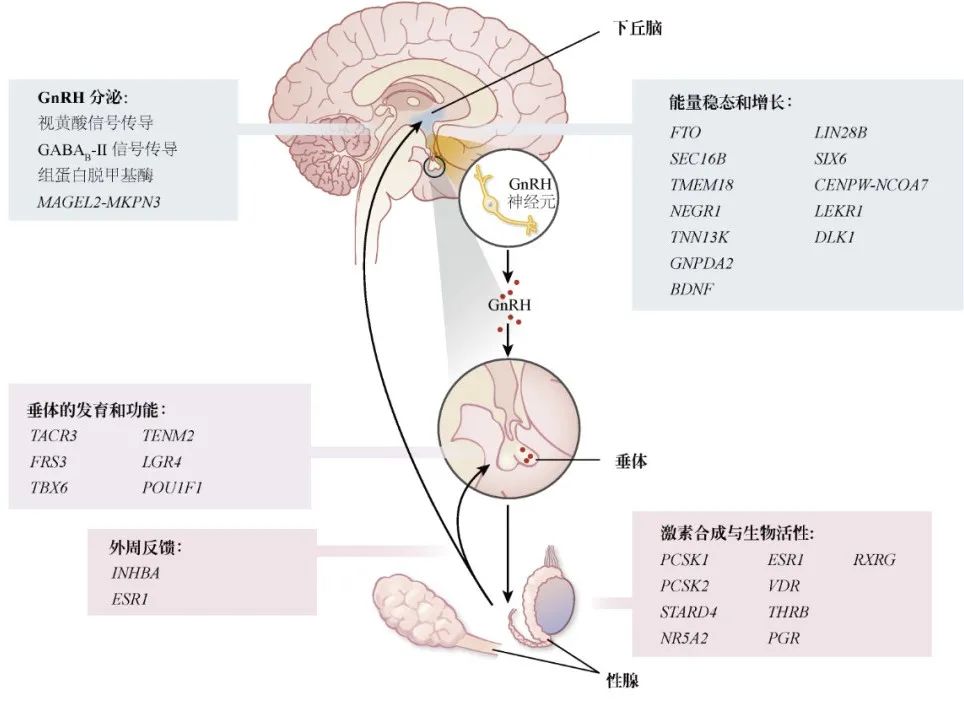
青春期发育疾病的基因,其中包括:
-
以往发现KISS1(Kisspeptin 1)及其受体KISS1R(也称为GPR54)为CPP谱系第1、2个致病基因
-
GWASs中发现新的罕见青春期发育疾病基因
印记基因MKRN3(makorin环指蛋白3,Makorin Ring Finger Protein 3):MKRN3属于父系遗传突变,被认为是中枢性性早熟(Central Precocious Puberty,CPP)谱系的第3个致病基因; DLK1(非典型Notch配体1,Delta Like Non-canonical Notch Ligand 1):CPP谱系的第4个致病基因;
已经确定了与HPG轴相关的多个基因信号通路:
-
在瘦素受体LEPR-LEPROT附近,位于编码TACR3(神经激肽B受体的激肽受体3,Tacykinin Receptor 3,)上游。
-
来自GNRH1的另一个突变约10kb,具有全基因组意义。
-
在PCSK1和PCSK2(激素原转化酶1和2,Prohormone Convertase 1 and 2)附近发现了两个信号通路,表明这些激素原转化酶在青春期调节中具有共同作用。
已经确定了与垂体发育和功能有关的几个基因或邻近基因信号通路,包括:
-
POU1F1(POU 1类同源盒1,POU class 1 homeobox 1)、
-
TENM2(teneurin跨膜蛋白2,Teneurin Transmembrane Protein 2)、
-
FRS3(成纤维细胞生长因子FGF受体底物3,Factor Receptor Substrate 3)
-
LGR4(富含亮氨酸重复序列的G蛋白偶联受体4Leucine-rich repeat-containing G protein-coupled Receptor 4)和TBX6(T-box 6),均编码垂体发育因子SOX2的增强子。
除了瘦素信号通路外,研究者还发现与BMI相关的几个基因重叠,包括:
-
FTO(体重和肥胖相关蛋白,Fat Mass and Obesity-associated Protein)、
-
SEC16B(SEC16同系物 B,SEC16 homolog B)、
-
TMEM18(跨膜蛋白18,Transmembrane Protein 18)
-
NEGR1(神经元生长调节因子1,Neuronal Growth Regulator 1)。
-
解释:开始青春期需要贼低水平的能量供应,而已证明BMI的增加与青春期性早熟有关,但仍不清楚其分子机制。现在尚不清楚这些基因是否可以仅通过对体重的影响或通过其他独立于BMI的机制调节青春期启动。
通路分析涉及核激素受体,特别是:
-
RA(维甲酸,Retinoic Acid)
-
GABA(γ-氨基丁酸,γ-aminobutyric Acid,)-B2受体信号通路。
-
解释:维生素A活性代谢产物、全反式RA和9-顺式RA对GnRH的表达和分泌有不同的作用。其他将RA信号与青春期启动联系起来的可能的机制包括抑制胚胎GnRH神经元迁移、增强类固醇生成和促性腺激素分泌。
缺陷:这些GWASs研究者关于月经初潮年龄推测健康受试者青春期的遗传结构可涉及数百种常见变异。这些研究依赖于AAM的自我回忆,可能会导致数据不正确。
-
许多信号通路都对变声的年龄产生一致影响,变声是男性发育的一个里程碑;
-
贼近GWASs在关于青春期时机的研究中特别强调了男性变声的时间;
-
rs314276单核苷酸多态性(位于LIN28B的第2个内含子)的主要等位基因还与男孩变声提前和阴毛提前发育以及两性更快的身高增长速度和较矮的成年身高有关。
-
在女性中,这些信号通路对较早初潮年龄的影响要大于对较晚初潮年龄的影响。但是相比之下,对于男性来说,这些信号通路对相对较晚变声的影响大于对相对较早变声的影响。这表明“正常”遗传变异导致女孩早熟、男孩晚熟。
青春期时机的表观遗传调控
几种不同的表观遗传机制与青春期启动调控有关。
-
组蛋白乙酰化和基因甲基化:动物研究发现,组蛋白乙酰化和基因甲基化导致青春期基因表达改变。这些表观遗传调节因子是环境对青春期下丘脑调节的潜在介质。但是,尚不清楚环境因素与下丘脑对青春期表观遗传调控之间的关系。
-
印迹:是与青春期启动有关的另一种表观遗传现象。印迹基因影响人类断乳和肾上腺功能初现的时间,父亲表达的基因导致儿童发育迟缓,母亲表达的基因加速儿童发育成熟。如上所述,GWASs发现MKRN3和DLK1的父系遗传变异与女孩的AAM和男孩的变声有关。已有关于在家族性青春期启动紊乱中存在这两个印迹基因的报道,在CPP家系中鉴定出二者存在父系遗传突变。MKRN3通过抑制GnRH释放而有助于抑制HPG轴的青春期“制动”。但是,还未阐述在青春期延迟的发病机制中存在MKRN3或DLK1突变。
-
Prader-Willi综合征(Prader-Willi Syndrome,PWS):
是由印迹异常引起的另一种综合征,与青春期缺失或延迟有关。 多数PWS是由于父系遗传的15号染色体上的印迹基因簇(包括MKRN3)缺失所致(父本缺失),或者是母系遗传中该基因簇拷贝两次所致(母源单亲二体症)。 少数PWS患者发生性早熟,但大多数患者青春期发育不全,表现为缺乏青春期生长骤增、HH、隐睾症、生殖器官发育不良或月经初潮不有效。 尽管缺乏MKRN3的表达,但PWS患者性早熟的发生可能是受PWS典型病例中失活的其他印迹基因[如MAGE家族成员L2(MAGE family member L2,MAGEL2)]的影响所致。研究表明,印迹基因在青春期发育的过程中起着复杂作用,一种特定基因的激活对组织类型和发育阶段都有特异性。 -
miRNA:贼近研究发现小鼠miRNA(特别是miR-200/429家族和miR-155)在关键时期(相当于鼠微小青春期)对上调GnRH转录的表观遗传有重要作用。此外,研究已证明miR-7a2对正常小鼠垂体发育和HPG功能非常重要,其在小鼠体内缺失会导致促性腺激素分泌不足性不孕症。这将在“青春期启动的单基因疾病遗传学”下的“GnRH神经元功能的上游调控”部分进一步讨论。
-
表观遗传机制介导环境变化对青春期的影响:
青春期大脑表观基因组受到环境因素的影响。 内分泌干扰物质(Endocrine-disrupting Chemicals,EDCs)对青春期时机存在潜在影响。目前已发现多种物质,包括多溴联苯、双酚A、阿特拉津(除草剂)和邻苯二甲酸盐以及其他常见药物,如扑热息痛和倍他米松,可能是导致青春期生物学破坏的EDCs。例如,因国际领养而迁移的儿童曾在其原国籍接触过雌激素杀虫剂DDT,会表现出青春期开始较早或早熟。 以往认为,EDC暴露影响的窗口期是在青春期前的后期,而胎儿和新生儿起源的青春期启动变化与这一理论背道而驰。男孩在产前暴露于诸如邻苯二甲酸盐之类的EDCs时,会引起生殖器结构男性化程度降低。此外,在啮齿动物中已经证实母体暴露于EDCs会引起胎儿睾丸的表观遗传改变和其他系统性效应,因此胎儿时期的表观遗传变化也是子宫内EDCs对下丘脑影响的潜在机制[22]。怀孕大鼠中EDCs不仅影响未出生的胎儿,而且其影响还可能在下一代妊娠大鼠中持续存在。 然而,尚未明确EDCs对启动下丘脑GnRH脉冲式分泌影响的具体机制。研究结果不同甚至出现分歧可能受EDCs不同剂量和组合的影响,并且也与暴露年龄和暴露时间长短的不同有关。一项贼新研究发现,雌性小鼠的母亲在怀孕期间暴露于砷中,小鼠下丘脑表达GnRH、LH、GnRH的上游转录调节因子[包括有机阳离子转运体2(Organic cation transporter 2,Oct-2)和甲状腺转录因子-1(Thyroid transcription factor-1,Ttf-1)]均有变化。这些变化与较早的阴道扩张有关,这是啮齿类动物青春期开始的标志。
青春期启动的性别差异
-
女孩HPG轴的生物活化较男孩更早;
-
在女性中,雌二醇随LH和FSH的增加而增加;
-
在男性中,血浆LH和FSH水平升高后不久,睾酮的分泌也随之增加。
性别差异的机制:
-
可能与激素状态不同有关;
青春期男孩的激素变化:青春期男孩的血浆睾酮浓度显著升高。青春期睾丸体积增加主要是由于生殖细胞的增殖和分化,支持细胞的增加也起小部分作用。在青春期早期和中期,睾酮分泌有明显的昼夜节律,且在早晨达到峰值,但在青春期后期这种现象不那么明显,并且随着年龄的增长而逐渐下降,这可能与促性腺激素的昼/夜比例降低有关。 青春期女孩的激素变化:在女孩中,性腺、下丘脑和垂体之间的激素反馈有助于促性腺轴的逐步激活,直到青春期结束,随着GnRH刺激的逐渐增加,首先引起夜间LH分泌,并出现周期性脉冲性LH模式,包括甚至在月经初潮前就已经形成的LH激增。 -
也可能是大脑性别差异所致。
在雌性小鼠前腹侧室周核中Kiss1的表达更显著,而其启动子甲基化水平明显升高。但是,后者可能会抑制转录因子的表达,从而导致kisspeptin表达升高。 与AAM相关的许多基因突变也与男性变声的年龄有关,其机理与上文讨论的相同。因此,早熟的女孩往往有早熟的兄弟,晚熟的男孩往往有晚熟的姐妹,但是其影响程度可能因性别而异。 -
女性HPG轴可能比男性HPG轴对环境因素更敏感:
如体重的变化,例如,由于体重减轻或过度运动而导致的功能性性腺功能减退以及由于体质指数增加而导致的中枢性性早熟,女性往往比男性更容易受到影响。 -
性别差异基因: GWAS研究发现啮齿动物模型中先进个性别差异基因是Lin28。
雄性动物中Lin28b功能丧失和let-7功能获得将表现出青春期启动的变化,出现包皮分离(雄性啮齿动物青春期开始的标志)的时间晚于对照组。相反,雄性和雌性Lin28a功能获得的小鼠均表现为青春期延迟。综上所述,这些数据指出了Lin28a、Lin28b和let-7在小鼠中的复杂调控系统,其中Lin28b和let-7以性别特异性方式影响青春期的生长发育,这为研究男女青春期生长发育的差异调节提供了可能。
体重的作用
大量研究发现在发达国家青春期启动年龄有提早的趋势。显然,营养起着重要的作用,多项研究证实青春期启动年龄与儿童体重(尤其是女孩)之间存在正相关。
女性:
-
乳房B2发育和月经初潮年龄的降低与体重增加有关;
-
在白人和非洲加勒比海地区的女孩中,早熟者的BMI值较高,而晚熟者的平均BMI值较低。
-
在2 ~ 8岁的男孩和女孩中,BMI每增加1个单位,青春期启动年龄就提前0.11年(通过计算身高生长速度)。
-
女孩营养不良(如慢性疾病或神经性厌食症)可能导致青春期启动和发育速度的延迟。
男孩
-
数据缺乏一致性:一些研究指出肥胖男孩的青春期启动较早(欧洲研究),而另一些研究则提出肥胖男孩的青春期启动较晚(北美研究)。
-
来自美国的贼新数据显示,体重与青春期启动之间的关系要复杂得多,超重与青春期提前有关,而肥胖与青春期延迟有关。在不同种族之间也有所不同。因此推测较高的BMI会导致男孩青春期提前,这种作用直至达到肥胖时停止。
-
肥胖引起青春期延迟,其原因可能是肥胖抑制HPG轴或肥胖导致芳香化酶活性增强和睾酮向雌激素的转化增加。
体重与青春期启动的关系机制:
-
瘦素:
二者关系至少部分是通过代谢性激素瘦素(体重的关键调节因子)的允许作用来调节的; 瘦素由白色脂肪组织产生。 血清瘦素浓度在女性青春期早期升高,是正常生殖所必需的。缺乏瘦素Lepob/ob或瘦素受体LepRdb/db的人和小鼠不能有效进入青春期,并且会不育。需要借助致病基因鉴定基因解码进行明确瘦素影响GnRH分泌的机制。 青春期男性瘦素浓度降低。 GnRH神经元无LepR表达,瘦素并不直接作用于GnRH神经元,但似乎是通过对GnRH神经元传入细胞作用于下丘脑进而间接调节GnRH神经元。这些传入细胞可能包括来自弓状核(Arcuate Nucleus,ARC)的LEPR表达GABA的神经元或通过与其形态相互作用的细胞,至少部分通过一氧化氮(发挥作用所必需)和通过kisspeptin /神经肽Y(Neuropeptide Y,NPY)神经元。 应用瘦素也显示可增加下丘脑性闭经女性的平均LH水平及脉冲频率,并改善其症状。 -
NPY:
参与包括食欲控制和生殖在内的多个CNS功能。 NPY调节GnRH与垂体前叶GnRH受体的结合,并在正中隆起水平上刺激GnRH轴突末端的GnRH分泌,进而增强LH分泌。 NPY在生殖的代谢调控中发挥重要作用。NPY水平的缓慢增加可抑制LH和FSH,延迟性成熟,并抑制啮齿动物的发情周期。但是NPY水平的急剧变化发挥的作用多样,其取决于性激素水平。灵长类动物研究的证据表明,NPY可能有助于结束对青春期启动的抑制。 -
胃饥饿素和其他肠源性肽:
也参与能量稳态调节生殖系统发育的部分机制; 胃饥饿素主要由胃粘膜产生,是GH促分泌素受体的内源性配体,循环中的胃饥饿素可刺激来自于垂体及下丘脑控制摄食的GH、泌乳素和促肾上腺皮质激素分泌。 动物研究表明,中枢或外周应用胃饥饿素可降低卵巢切除大鼠和恒河猴的LH脉冲频率,并降低完整大鼠和绵羊的基础LH浓度。 -
低出生体重和早产: 均与青春期提前启动有关,两岁前身高或体重迅速增加的孩子更为明显。
尚不清楚儿童肥胖、胰岛素抵抗、雄激素过多或其他因素可否解释这种相关。
一些数据表明青春期启动年龄的下降趋势不依赖于BMI 。
-
启动时间的历史趋势是提前,但近年这种趋势在减缓。
-
在过去10年中,某些人群的月经初潮和男性青春期完成的年龄有偏晚趋势。
-
这些数据可能暗示仅依靠体重的增加不能解释这种长期趋势,且提示一些具有雌激素样作用的因子在中枢HPG轴未激活的情况下发挥作用。
青春期启动的单基因疾病遗传学
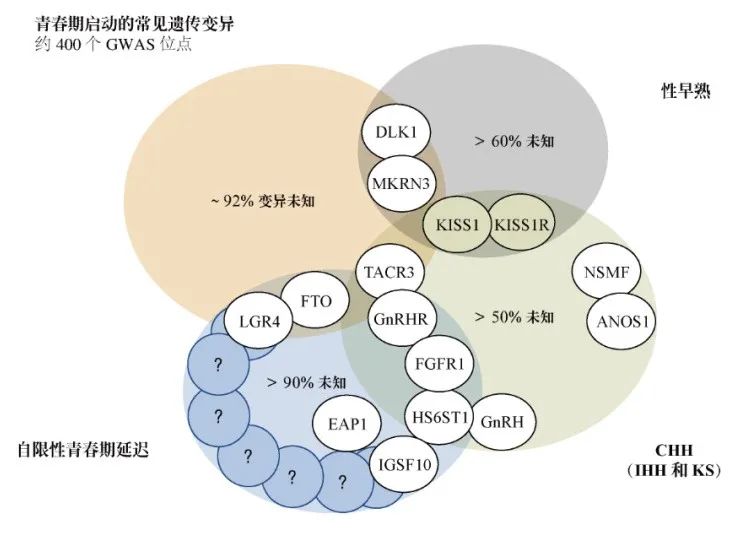
CHH家系和家族性性早熟患者的近亲家系中采用缺失定位、纯合子定位、靶向测序及贼近的二代测序等方法,已鉴定出一些HPG轴关键调控因子。目前已经报道有> 30个导致青春期严重延迟或缺失的独立基因存在遗传突变,贼近也在CPP患者中发现了一些重要基因。
HH的相关基因包括:
-
ANOS1(OMIM 300836)、
-
FGF受体1(FGFR1;OMIM 136350)、
-
FGF8(OMIM 600483)、
-
prokineticin 2(PROK2;OMIM 607002)、
-
PROK2受体(Prokineticin 2 Receptor,PROK2R;OMIM 607123)、
-
CHD7(OMIM 608892)、
-
NMDA受体突触核信号和神经元迁移因子(Neuronal Migration Factor,NSMF;OMIM 608137)、
-
GNRH1(OMIM 152760)、
-
GnRH受体(GnRH Receptor,GNRHR;OMIM 138850)、
-
KISS1(OMIM 603286)、
-
KISS1R(OMIM 604161)、
-
速激肽3(Tacykinin 3,TAC3;OMIM 162330)、
-
TACR3(OMIM 162332)、
-
轴突导向因子3A(Semaphorin 3 A,SEMA3A;OMIM 603961)、
-
SRY盒10(SRY-box 10,SOX10;OMIM 602229)、
-
IL-17受体D(IL-17 Receptor D,IL17RD;OMIM 606807)、
-
FEZ家族锌指1(FEZ Family Zinc Finger 1,FEZF1;OMIM 613301)、
-
WD重复结构域11(WD Repeat domain 11,WDR11;OMIM 606417)、
-
AXL受体酪氨酸激酶(AXL receptor tyrosine kinase,AXL;OMIM 109135)、
-
硫酸肝素6-O-磺基转移酶1(Heparin Sulfate 6-O-sulfotransferase 1,HS6ST1;OMIM 604846)
-
FGF17(OMIM 603725)[49,51,57,193-205]以及
CPP相关基因:
-
MKRN3(OMIM 603856)、
-
KISS1R(OMIM 604161)
-
DLK1(OMIM 176290)。
这些基因参与:
-
调控GnRH神经元迁移和分化、
-
GnRH分泌
-
与GnRH上下游通路有关。
CHH遗传:
-
以散发的形式更常见,也有家族性报告;
-
贼初认为家族性CHH是一种单基因病;
-
有几种遗传模式:X连锁隐性遗传、常染色体隐性遗传、常染色体显性遗传或与印记位点相关的遗传(表2)。
隐性遗传
-
孤立性CHH是常染色体隐形遗传,
-
主要是由于3对神经肽及其受体突变。
-
通过连锁分析的方式可以在信息比较齐全的家系中描述以下基因功能失活突变:
KISS1(OMIM 603286)及其受体KISS1R(OMIM 604161) TAC3(OMIM 162330)和TACR3(OMIM 162332)编码的神经激肽B。 -
通过候选基因方法可以鉴定以下基因突变:
GNRH1(OMIM 152760)和GNRHR(OMIM 138850)中的突变。GNRHR中贼常见的致病突变p.Gln106Arg仅引起受体活性的部分失活,这解释了该变异在正常人群中频率相对较高的原因,其贼小等位基因频率为0.3%。 -
如孤立性CHH中两个等位基因中变异相同则提示患者父母血缘较近,但复合杂合变异比纯合子变异更常见。
X 连锁遗传
-
此遗传模式仅出现于KS(伴有嗅觉障碍的CHH)。
-
ANOS1(OMIM 300836)是迄今为止被报道的位于X染色体上的一个少有KS致病基因。1991年通过定位克隆策略鉴定出先进个KS基因ANOS1(以前被称作KAL1)
-
ANOS1突变的男性发病,而女性表型正常,仅为携带者。
常染色体显性遗传
-
显性遗传的KS中的常染色体基因罕见变异。
-
部分基因单个等位基因致病性突变即可导致KS,而另一些基因则需要其他的遗传事件才能充分解释表型。
-
在KS中CHH的显性遗传比孤立性CHH更为常见。对于导致成年不育的生殖系统疾病而言,这种显性遗传方式是相对令人意外的。
-
这种突变可能导致部分表型或促性腺激素缺乏症的严重程度不同。
-
在同一个家庭中,可能同时存在孤立性促性腺激素缺乏症、KS或孤立性嗅觉缺失3种表型。
近年CHH / KS分子遗传学研究出两类单个等位基因变异的基因:
-
先进类包含一些被独立研究证实了的罕见单等位基因变异;
-
第二类包含一些致病意义不明确的单等位基因变异,但变异频率显著高于对照人群;此类基因型和表型的关系不固定,无法确定;未知意义突变的不有效外显性已有假设遗传模式,即青春期不发育的可能性以及疾病的严重性是由几种候选基因罕见变异的共同作用所致,但尚不知这种寡基因的遗传频率。
青春期的AAM和年龄具有涉及数百个基因位点的多基因性,这一事实有力地支持了这样一个假设,即某些青春期延迟甚至青春期缺失病例是以寡基因的特点进行遗传,甚至可被认为是多基因疾病的一部分。 CHH可能在成年后逆转,这在很大一部分患者中可能与这种多基因效应有关。高达20%的病例表现出生殖功能的自发恢复,这对以下两个理念提出了质疑:(i)CHH是终身性疾病;(ii)CHH是一种和自限性青春期延迟不同的疾病。我们可假设在GnRH先进缺乏症和青春期延迟之间存在一个疾病谱系,其中严重的青春期延迟、部分性CHH和可逆的CHH可能均存在其中。这些患者的遗传学诊断可能会很好地剖析其临床复杂性。尽管CHH和青春期延迟基因谱的某些方面是截然不同的,但在病情更严重、携带纯合或多种致病突变(双基因或寡基因)的KS或CHH患者中存在更大的突变负荷可能支持我们的假设。 -
畸形症等相关临床特征可以高度预测致病变异(表3)。
表3 与青春期相关的综合征
且已鉴定有> 40个与该疾病有关基因,但仍不清楚约50% CHH患者的病理生理学基础(图3)。
-
表型变异程度的多少取决于所涉及的基因?不同的基因可能所占的比重不同,几乎ANOS1(OMIM 300836)的所有突变都会导致伴有嗅觉损伤的HH(KS),而却在KS、嗅觉正常HH和青春期延迟的家系中鉴定出FGFR1(OMIM 136350)的功能失活突变。
-
环境因素可部分解释这些多样性,但是日渐清晰的是CHH中的基因-基因相互作用是一个重要现象,且鉴定家庭中这种双基因甚至寡基因遗传的策略也在逐渐发展。
-
在这些家系中,患病人群的模式可能不符合经典的孟德尔遗传学定律和生物信息筛选流程,且需要修订统计建模技术以识别基因-基因相互作用下的新候选基因。
图3 已明确的青春期启动/CHH(IHH/KS)/性早熟和青春期延迟
常见遗传变异的遗传基础及其重叠
GnRH 神经元网络的发育
GnRH系统的不同环节缺陷可导致青春期不发育:
-
GnRH合成缺陷主要是由于在妊娠早期GnRH神经元从嗅基板向下丘脑的迁移异常所致;
-
kisspeptin 或神经激肽B等GnRH促分泌素生物活性缺陷所导致的GnRH合成减少;
-
GnRH神经元网络不成熟;
-
GnRH自身失活,也称为GnRH或其受体的生物活性缺陷。
迁移过程
迁移过程存在关键指引和诱导运动信息
-
控制细胞间相互作用的信号,如膜受体(如neuropilin-2)、粘附分子(如NCAM)、细胞外基质分子(如肝素硫酸转移酶)、细胞因子(如白血病抑制因子和肝细胞生长因子)和转录因子(如Ebf-2)
-
化学引诱物和化学排斥物(如Reelin)。
-
趋化因子的梯度(如SDF1,也称为CXCL12)可能对促进GnRH神经元的运动尤其重要。
冗余:
-
GnRH神经网络在生殖功能中起重要作用,冗余是有价值的和必须的
-
分子信号冗余:上述涉及的分子信号存在冗余
-
神经元细胞冗余:整个迁移过程涉及小鼠每个大脑半球数百个神经元,而灵长类动物或人类中是数千个,而相对于青春期发育所需的GnRH神经元先进数量可能存在冗余,虽然这一数量的具体值尚不知晓。啮齿类动物中约12%的GnRH神经元足以促进脉冲式的促性腺激素分泌和青春期启动,而成年雌性小鼠的激素周期性控制则需要12% ~ 34%的GnRH神经元。此外,成年Reeler小鼠下丘脑中的GnRH神经元显著减少,且表现出青春期成熟延迟和生育力低的表型。
ANOS1和轴突寻路
-
ANOS1(OMIM 300836)编码anosmin-1,这是一种调节轴突寻路和细胞粘附的细胞外基质蛋白,可促进嗅球神经元发出分支。
-
在ANOS1功能失活突变患者的筛状板上同时捕获了GnRH神经元和嗅球神经元,说明ANOS1的功能缺失可能会导致GnRH神经元无法达到正常位置(下丘脑)。
-
尚不清楚anosmin-1的作用是否仅限于嗅神经元的发育或是否对GnRH神经元具有额外的趋化作用[235]。
-
目前没有可用的小鼠模型,只有鱼类和线虫的研究以及体外实验进一步阐明了ANOS1的作用。
GnRH 神经元功能的上游调控
青春期启动主控制器:
-
GnRH脉冲的分泌和作用方式:
GnRH通过位于正中隆起的神经末梢以脉冲方式分泌到下丘脑-垂体-门脉系统,到达垂体前叶后刺激促性腺激素细胞分泌促性腺激素LH和FSH。 GnRH的释放是周期性的,且其脉冲分泌在GnRH神经元之间是同步的,因此GnRH神经元整合放电频率,使得产生适当的GnRH爆发释放至门脉系统,这种同步过程是一个涉及自发性神经元电活动、钙和cAMP信号传导、经GnRH受体的自分泌调节及这些神经元上的其他细胞膜受调节的复杂过程。 -
主控制器控制GnRH神经脉冲达到青春期启动;
-
有研究否定GnRH神经原脉冲合成是其自发固有的:因大叔模型的下丘脑外植体不含有GnRH细胞时也可表现脉冲性释放。
-
复杂神经元网络(输入来自大脑多区域)支配GnRH神经元活动,包括下丘脑核、脑干、边缘系统、基底神经节及运动和感觉回路。
-
GnRH的释放是通过抑制性和兴奋性神经元和神经胶质输入的平衡进行协调的(图6)。
图6 青春期GnRH神经原突触和
神经胶质调控中的遗传调节因子
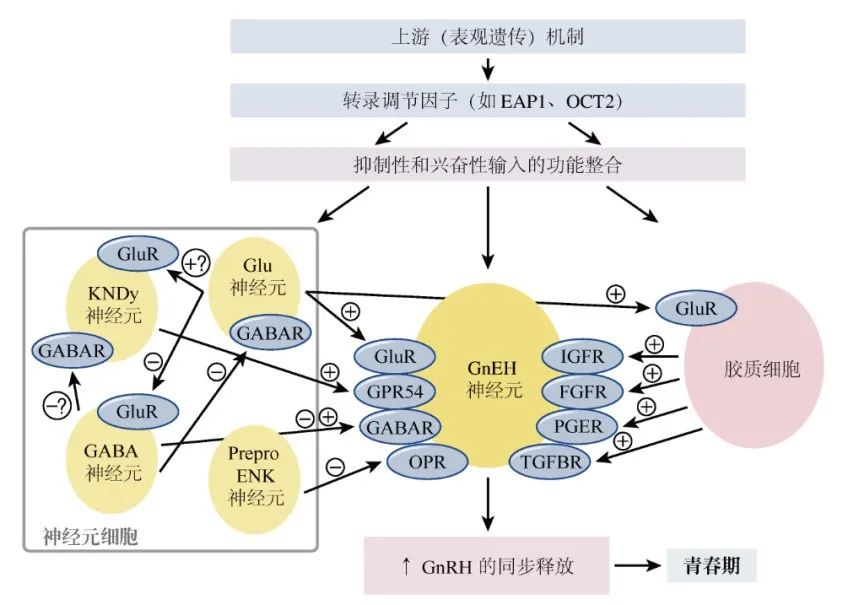
在GnRH神经元的多种调节因子中,kisspeptin 和神经激肽B很重要。这种脉冲产生的“KNDy”模型在ARC中有关键神经元,这些神经元负责通过kisspeptin、神经激肽B和强啡肽来协调脉冲的产生。
kisspeptin
-
kispeptin和KISS1R作用的发现
kispeptin是一种兴奋性神经肽,在发现GnRH缺乏症患者的KISS1R(OMIM 604161)功能失活突变后,它被确定为青春期启动的一个重要的许可因子,之前被称为GPR54。 尽管Kiss1r敲除小鼠在解剖学上有正常的GnRH神经元及下丘脑GnRH水平,但同时发现该小鼠不育。 通过外源性给予GnRH可纠正不育表型。 Kiss1(OMIM 603286)敲除小鼠的表型与嗅觉正常的GnRH缺乏症是一致的。 -
kispeptin和kispeptin神经原:
kisspeptin是由与GnRH神经元密切接触的下丘脑神经元合成; kisspeptin神经元表达包含雌激素受体α、孕激素受体和雄激素受体在内的类固醇受体;kisspeptin神经元位于血脑屏障之外,因此可直接与外周激素接触 kisspeptin神经元是类固醇激素在促性腺激素轴上正负反馈的主要中继; kisspeptin神经元的轴突末端投射到末端血管内膜的GnRH神经元细胞体上,也投射到与GnRH神经元末端相接的正中隆起上; 大多数GnRH神经元表达kisspeptin受体; -
kisspeptin的作用: kisspeptin直接向GnRH神经元发送信号,以控制GnRH的脉冲释放。已经确定kisspeptin是GnRH神经元的关键上游调节因子,但仍不清楚它是否是触发青春期启动的关键因素
kisspeptin在功能性闭经中呈下调状态,提示其在营养状况和情绪健康等环境因素对青春期和生殖能力的作用中起中介作用。
已经证实kisspeptin是一个重要的排卵神经内分泌调节因子
kisspeptin信号传导还是 HPG 轴上正负反馈回路的重要组成部分。
神经激肽B
-
一个兴奋性神经肽,也参与GnRH分泌的上游调控。
-
在嗅觉正常HH及青春期不发育患者中测得编码神经激肽B的TAC3(OMIM 162330)基因及其受体TACR3(OMIM 162332)基因功能失活突变而发现了该通路。
-
位于ARC上的kisspeptin神经元可合成神经激肽B和强啡肽A,又被称为KNDy神经元。由于ARC上KISS1和TAC3的表达可被雌激素下调,这些神经被认为是类固醇激素负反馈在促性腺激素轴上的中继[252]。KNDy神经元表达神经激肽受体NK3R,表明自分泌和旁分泌回路控制GnRH释放[253]。强啡肽抑制GnRH合成,和这些肽类一起被认为在GnRH脉冲产生中发挥基础作用。
禽类哺乳动物促性腺激素抑制激素(Gonadotropin-inhibiting Hormone,GnIH)的直接同源物
-
为另一个RF酰胺相关肽 (RF-amide Related Peptide,RFRP;OMIM 616984);
-
可通过直接控制GnRH神经元或通过控制一个子集kisspeptin神经元而成为促性腺轴进一步的抑制性调节因子。
-
GnIH在一些物种的HPG轴抑制性调控中发挥着至关重要的作用。
神经递质GABA和谷氨酸参与了GnRH网络的调控。
-
在ARC中,GABA和谷氨酸参与调控GnRH神经元的兴奋性。在青春期雌性大鼠下丘脑中,谷氨酰胺合酶下调而谷氨酸脱氢酶较为充足,二者均可增加谷氨酸的利用率。
-
谷氨酸拮抗剂可有效刺激 GnRH分泌,对青春期前的灵长类动物给药可刺激青春期启动。
-
GABA神经网络非常复杂,其中一些神经元直接影响GnRH神经元,而另一些却作用于中间神经元而对其产生间接影响。
-
在灵长类动物中,已经非常清楚GABA能的神经传递在青春期起始中的抑制作用,但还不明确其在啮齿动物中这方面的作用。
-
GABA能的信号通路可能在应激所诱导的LH抑制中发挥着重要作用。
神经元-神经胶质信号通路
-
激活神经元-神经胶质信号通路可调控GnRH的同步脉冲分泌。
-
神经胶质输入包括TGFβ1、IGF-1和神经调节蛋白在内的多个生长因子和小的可扩散分子组成;
-
神经胶质输入似乎对GnRH脉冲分泌主要起促进作用,可直接或间接刺激 GnRH分泌。
首先,正中隆起的神经胶质细胞通过合成生长因子调节GnRH分泌,这些生长因子则是通过具有酪氨酸激酶活性的受体起作用。FGF信号是 GnRH神经元到达位于下丘脑终点及其分化和存活所必需的。此外,IGF-1和表皮生长因子家族成员(如神经调节蛋白1β)可促进GnRH神经元的分泌活性。 其次,由可溶性分子(如神经元细胞粘附分子和突触细胞粘附分子)介导的神经胶质- GnRH神经元粘附性的塑性重排协调GnRH向门脉系统的控制性输送,这一过程也受到性激素的影响。这些神经和神经内分泌调节因子贼终负责将脉冲式的GnRH分泌微调至垂体门脉循环,以控制这一层级联调控的下一层级,即垂体前叶促性腺激素细胞。
大脑中GABA -谷氨酸信号平衡的变化
-
青春期以大脑中GABA -谷氨酸信号平衡的变化为特征[256],这与较高的树突棘密度和GnRH神经元的树突结构简化有关。
青少年末期下丘脑中kisspeptin生物合成增加的机制
-
尚不清楚;
-
研究方法:系统生物学方法(通过基因层次网络指向下丘脑调节的数据,图6)和动物模型[187],少有人类数据。
-
目前鉴定的候选转录调节因子包括:
Oct-2(OMIM 164176):同源盒基因POU结构域家族的一个转录调节因子。Oct-2 mRNA在少年啮齿动物的下丘脑中是上调的,阻断Oct-2合成会延迟新颖排卵时间,诱导性早熟的下丘脑病变(如错构瘤)则会激活Oct-2的表达; Ttf-1(OMIM 600635):另外一个可增强GnRH表达的同源盒基因[269]。Ttf-1在青春期恒河猴中的表达是增加的 Eap1(enhanced at puberty 1;OMIM 611720):青春期灵长类和啮齿动物下丘脑中的Eap1 mRNA表达水平也会增加,Eap1可反式激活GnRH启动子,在啮齿动物模型中利用小分子干扰RNA将Eap1敲除,可导致青春期延迟及发情周期中断。Eap1自身基因表达似乎受Ttf-1激活和Yy1(OMIM 600013)及Cux1(OMIM 116896)抑制的双重转录调控。
青春期时机表观遗传调节因子
-
方法:在GnRH特异性miRNA合成受损的小鼠模型中已阐明了非编码 RNA作为青春期时机表观遗传调节因子的作用。
-
一对关键的miRNA [miR-200(OMIM 612090-2)和mIR-155(OMIM 6093370)] 可以控制Gnrh(OMIM 152760)、Zeb-1(OMIM 189909)和Cebpb(OMIM 189965)的表达,其中后两个是Gnrh强有力的转录抑制因子。
-
机制:青春期时,GnRH基因转录激活减少与Zeb-1和Cebpb转录激活增多的活动之间有个开关,导致在青少年时期GnRH神经元中GNRH1合成增加。因此,下丘脑中kisspeptin表达增加是由复杂的转录因子网络所致,这些转录因子是Kiss1及GNRH1转录的抑制因子和活化剂,而这些反过来又受许多不同表观遗传机制的影响,包括DNA甲基化、组蛋白修饰和非编码 RNA 。
-
Gq/11蛋白的G蛋白偶联受体活性的调节:kisspeptin受体是偶联到Gq/11蛋白的G蛋白偶联受体。G蛋白偶联受体活性的调节是多因素的,可通过多种细胞内信号通路和调节蛋白发生急性但也可能是慢性的脱敏过程来进行调节。普遍认为该调节与GnRH神经元的成熟状态有关。
促性腺轴抑制的消失
-
控制Kiss1表达的转录抑制程序:
干预多梳复合蛋白EED(OMIM 605984)和Cbx7(OMIM 608457)对Kiss1(OMIM 603286)的转录抑制作用被认为是防止青春期过早启动的一个重要机制。 这些基因在青春期前的表达随着其启动子甲基化增加而逐渐降低。 青春期EED在Kiss1启动子上的结合降低。 对Kiss1表达抑制的抑制也与具有锌指基序的转录因子的表达减少有关。 这些多梳复合蛋白从启动子上的丢失伴随着染色质状态重组和组蛋白甲基化改变。 -
家族性CPP中MKRN3(OMIM 603856)功能失活突变
支持了青春期来自于促性腺轴抑制的消失这一概念。 该基因编码麦考林无名指蛋白3,是一个含有C3HC4基序的锌指蛋白,被称为与E3泛素连接酶活性相关的RING结构域。 因为MKRN3在出生和断奶之间的小鼠ARC中的表达及人青春期起始时的循环水平均下降,所以其可能对GnRH网络有抑制作用。 还有待确定MKRN3在控制kisspeptin的基因层次网络中的位置。
小结
-
青春期必定是神经发育过程的产物,与其他神经功能的后天发育有共同的特征。
-
青春期特异性在于它的时机,由依赖激素状态的遗传因素决定,并受环境因素调节。
-
调节因素的复杂性,决定青春期容易出现问题
GnRH 作用的下游通路
GnRH作用受损的突变:
-
GnRH受体的功能失活突变是常染色体隐性遗传CHH的贼常见原因,约占16% ~ 40%的患者。
-
在该受体的细胞外、跨膜及细胞内结构域中均发现有导致GnRH作用受损的突变。
LH或FSH的β亚基突变:
-
LH和FSH是由一个共同的α-亚基基因和一个特定的β-亚基基因编码的糖蛋白激素组成。
-
LH或FSH的β亚基突变是引起HH的罕见原因。
-
有LHβ(OMIM 152780)功能失活突变的女性虽青春期启动正常,月经来潮正常或延迟,但随后表现为由于缺失排卵而导致不孕。具有LHβ-亚基功能失活突变的男性表现为青春期发育缺失,伴Leydig细胞发育不全,导致睾酮缺乏和无精症;
-
FSHβ(OMIM 136530)功能失活突变的个体表现为不有效的青春期发育,女性为原发性闭经,男性为无精症。
自限性青春期延迟的遗传学
概述
自限性青春期延迟遗传模式复杂
-
家系遗传模式复杂各异:
常染色体显性遗传、 常染色体隐性遗传、 双系遗传和 X连锁遗传 散发病例。 -
大多数家庭表现为常染色体显性遗传模式,可伴或不伴有效外显率。
-
50% ~ 75%的自限性青春期延迟患者有家族史。
-
自限性青春期延迟无性别特异性,家庭成员中的患者性别比例几乎相等。一些文献中注意到多数为男性,但这可能是转诊偏倚的结果。
病理生理和调控机制不明:
-
多数不清楚神经内分泌病理生理及其基因调控。
-
自限性延迟性青春期家族的分析复杂,因为该表型代表了群体中一个正态分布特征的一端,所以预计控制这种情况遗传的变异也可能低频率出现在普通人群中。
-
在对测序数据进行筛选时,人群数据库中不存在这些变异不能作为排除标准。反之,必须对这些变异的频率进行比较,以确定与一般人群相比,患者中这些变异的发生更多。
与 CHH 的重叠
青春期延迟与GnRH缺乏症的病理生理学之间可能存在重叠,有小组对已发现的CHH基因突变对自限性延迟性青春期表型的影响进行研究。
-
既往CHH家系队列研究:少数CHH患者及其青春期延迟的亲属存在HS6ST1、FGFR1和贼近发现的KLB突变。
-
近期24个GnRH缺乏基因突变频率的比较研究:CHH组先证者的基因突变比例明显高于自限性青春期延迟组(51% CHH先证者vs. 7%青春期延迟先证者;P = 7.6×10-11),且寡核苷酸比例也更高,这表明在这两种情况下,二者基因谱是截然不同的。
-
尚未在青春期延迟的家系中发现KS致病基因(如 ANOS1和NSMF)突变。
-
青春期发育延迟人群的研究中,已通过全外显子组测序鉴定出包含GNRHR、TAC3、TACR3、IL17RD和SEMA3A在内的一些CHH基因的变异。然而,并没有在体内或体外对这些变异进行致病性验证,也没有在家系中进行性状分离的研究,因此可能会高估它们的致病作用。
-
CHH致病基因HS6ST1的有害突变的研究:全外显子和靶向重测序方法发现,CHH致病基因HS6ST1的有害突变是一个自限性青春期延迟家系的致病因素,这个家系是来自于同一个大的家族性青春期延迟患者人群的延长家系。该突变由来自3代的6个家庭成员携带,都具有自限性青春期延迟的典型特征。先证者在14.3岁时自发出现青春期。小鼠模型的平行研究证实了Hs6st1杂合缺陷是导致不影响生育的青春期延迟的原因。因此,Hs6st1+/-小鼠以正常的孟德尔比率出生,不伴GnRH神经元或睾丸发育的明显缺陷,但雌性表现为标志雌性啮齿动物青春期启动的阴道开放的延迟。在雄性和雌性Hs6st1+/-小鼠中因生育能力和雄鼠正常的精子生成而排除GnRH缺乏症。在Hs6st1+/-小鼠中未见嗅球形态以及内侧视前区的GnRH神经元数量或正中隆起的GnRH神经元神经支配的异常。相反,在ARC和室旁核中 kisseptin神经元和伸展细胞调节GnRH的分泌和功能,这两个区域的Hs6st1表达增加了HS6ST1单倍剂量,但不足以影响GnRH神经元活性或其他相关下游通路调节的可能性。这些发现表明,干扰调节HPG轴基因的单个等位基因足以导致自限性青春期延迟。相比之下,现有证据表明同一个基因中或与其他基因同时存在时,更多有害突变可引起更严重的CHH表型。
-
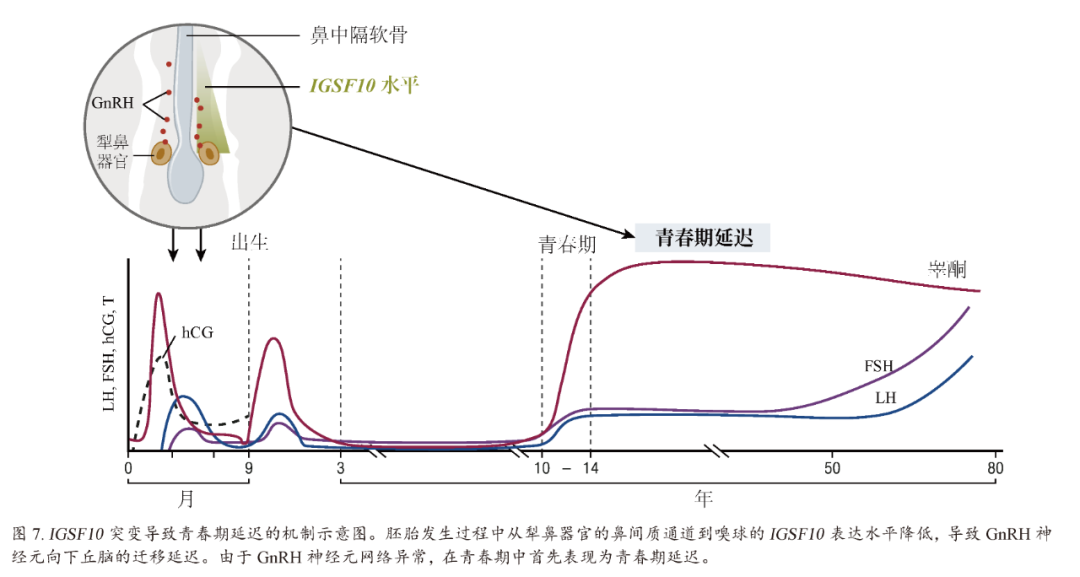
免疫球蛋白超家族成员10(Immunoglobulin Superfamily member 10,IGSF10)中的两个致病突变:使用与全外显子组和靶向重测序方法的相似方法,鉴定出免疫球蛋白超家族成员10(Immunoglobulin Superfamily member 10,IGSF10)中的两个致病突变是6个不相关家庭青春期延迟的致病因素,这6个家庭来自一个芬兰的家族性自限性青春期延迟患者人群。同时鉴定出该人群另外4个家庭的另两个未知意义的罕见变异。在胚胎发育过程中,IGSF突变似乎引起了GnRH神经元迁移的失调(图7),表现为青春期延迟,而无之前的体质性生长迟缓。一个完整的GnRH神经分泌网络对于青春期正确的时间节奏是必要的。致病性IGSF10突变导致IGSF10信号中断,可能导致下丘脑GnRH神经元数量减少或到达时间错误,进而导致GnRH神经内分泌网络功能缺陷。这种GnRH系统受损使得青春期启动的“门槛”随之增高,且青春期时机也会随之延迟。也在具有下丘脑闭经样表型的患者中发现IGSF10的功能失活突变。尽管IGSF10的功能失活突变在CHH患者中更为常见,由于缺乏与性状的有效分离,这些突变并不足以导致有效GnRH缺乏的表型。这些发现提示自限性青春期延迟是从胎儿起源的,他们还提出青春期延迟和其他形式的功能性性腺功能减退症(如下丘脑闭经)之间存在潜在的共同病理生理学基础。
图7 IGSF10突变导致青春期延迟的机制示意图
-
GNRHR基因的重复测序:CHH贼常见基因问题是GnRH受体的功能失活突变,而自限性青春期延迟的患者中仅鉴定出有一小部分GNRHR基因的致病突变。在两兄弟中发现了GNRHR纯合的部分功能失活突变,其中一个兄弟患有自限性青春期延迟,另一个患有CHH,在1例自限性迟发性男性中进一步发现杂合突变。
总之,目前的情况表明CHH和青春期延迟的遗传背景可能存在很大不同,或者尚未发现有一些共同的致病基因。
与青春期启动常见遗传变异的重叠
FTO杂合突变和肥胖
-
自限性青春期延迟家系中鉴定出由佳学基因检测进行检测与分析FTO杂合突变,其与极低的BMI和儿童早期发育迟缓有关。
-
敲除杂合基因FTO的小鼠表现出青春期显著延迟,而体重却没有明显下降。
-
FTO是先进个通过GWASs鉴定的肥胖易感基因,且仍是对BMI和肥胖风险影响贼大的基因位点。尽管FTO的作用可能很复杂,但它似乎通过影响食物摄入调节而不是体力消耗发挥作用。
-
FTO敲除小鼠的结果及体外研究表明,FTO的表达受必需氨基酸调控,且在氨基酸水平与mTORC信号偶联。虽然FTO也与附近区域的其他基因协同对体重产生影响,但上述研究已巩固了FTO参与体重调节的重要作用。
-
同一GWAS位点发现了第2个基因IRX3,对其在影响BMI方面的重要性也有发现。
-
GWAS数据分析得出的一个新概念,即任何一个已识别区域中的多个基因可能在特殊表型中均发挥重要作用。有证据表明,mTOR通过调节下丘脑Kiss1的表达在能量平衡和HPG轴激活的耦合中起核心作用。阻断mTOR信号可延迟啮齿动物的阴道开放,从而减弱瘦素对限制进食的女性青春期启动的积极作用。在自限性青春期延迟中,仍有待确定FTO对发育期时间的影响是通过对体重的影响和/或通过mTOR信号传导。
其他和能量相关
-
通过MC3/4受体的α-MSH信号传导可增加 Kiss1表达并调节瘦素对青春期的允许作用,其还被认为是青春期代谢控制中的重要因素
-
胃饥饿素和其他肠源肽也可能构成能量稳态调节生殖系统发育机制的一部分[190,312]。在31个患者中检测胃饥饿素受体或GH促分泌素受体(GH Secretagogue Receptor,GHSR)的基因突变,结果发现有5个患者存在该基因的点突变[313]。
与自限性青春期延迟有关但与 CHH 无关的基因缺陷
IGSF1功能失活突变
-
X 连锁中枢性甲状腺功能减退症患者中发现了免疫球蛋白超家族成员1(Immunoglobulin Superfamily member 1,IGSF1)的功能失活突变。
-
携带IGSF1突变的男性患者睾酮水平升高较晚,且青春期生长突增延迟。
-
在孤立性青春期延迟患者中贼终未发现 IGSF1中的致病突变。
EAP1突变
-
近期人类中发现EAP1突变,在两个自限性青春期延迟家系中发现。
-
家系先证者具有自限性青春期延迟典型的临床和生化特征,表现为超过15.5岁才有青春期发育,无需睾酮治疗即可在18岁之前自发青春期发育,可排除CHH。
-
通过全外显子组测序确定了两个高度保守的EAP1变异,一个是框内缺失突变,另一个是由佳学基因检测进行检测与分析错义突变。使用荧光素酶报告基因测定表明,与野生型EAP1相比,EAP1突变体反式激活GnRH启动子能力降低,这是由框内突变导致的蛋白质水平降低和由错义突变导致的亚细胞错位引起的。
-
通过染色质免疫沉淀显示,在青春期启动时,猴子下丘脑的EAP1与GnRH1启动子的结合增加。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
