












佳学基因导读
体外受精胚胎的植入前遗传学检测 (PGT) 是阻断遗传、降低遗传病在人群和家族中遗传中的一个有效方法;然而,目前仍缺乏更全面的胚胎遗传学评估,无法将常见变异和罕见变异的影响结合起来。在遗传病的阻断研究中,研究机构结合分子和统计学技术,可靠地推断了 110 个胚胎的基因组序列,并模拟了 12 种常见疾病的易感性。分析发现,在来自第 5 天胚胎活检的病例中,与多基因风险评分相关的位点的基因型准确率为 99.0% 至 99.4%,而在来自第 3 天胚胎活检的病例中,基因型准确率为 97.2% 至 99.1%。将罕见变异与多基因风险评分 (PRS) 相结合会放大同胞胚胎之间的预测差异。例如,对于携带致病性BRCA1变异的夫妇,阻断遗传课题组预测,当结合 BRCA1 或 PRS 时,兄弟姐妹之间的比值比 (OR) 差异为 15 倍,而单独使用BRCA1或 PRS时,差异为 4.5 倍或 3 倍。这一研究结果可能为基于基因组的 PGT 在临床实践中的实用性和实施的讨论提供参考。
关键词:预测医学、诊断标记、基因组学
结合亲本基因组的全基因组测序和植入前胚胎的基因分型的计算方法可以准确预测胚胎的遗传基因组并计算多基因风险评分。
以下是详细介绍
PGT 可以在植入前对胚胎进行家族特异性遗传疾病分析。虽然 PGT 目前用于预防罕见的孟德尔遗传疾病1、2 ,但有几个团队已经探索将检测范围扩大到常见疾病,如心脏病和癌症3-6。这些方法依赖于使用 PRS 7 ,它将数十、数百或数千种遗传变异的影响结合成一个预测因子。然而,由于单细胞或少细胞胚胎活检中 DNA 的数量和质量有限,全面分析胚胎基因组的尝试成本高昂且耗时8-10 ,存在与等位基因丢失相关的不准确性,需要远亲2或依赖于填补,这妨碍了检测BRCA1 11等基因中罕见的有害变异。为了克服这些限制,阻断遗传课题组扩展了全基因组重建 (WGR) 策略1 ,该策略使用亲本基因组测序和胚胎基因分型来预测胚胎的遗传基因组序列。在本文中,阻断遗传课题组将这种方法应用于10对夫妇的110个胚胎,并计算了12种疾病的多基因预测因子,包括癌症、心脏代谢疾病和自身免疫性疾病(图1)。阻断遗传课题组将这些胚胎的重建基因组和多基因预测结果与相应出生儿童组织样本生成的预测结果进行了比较。
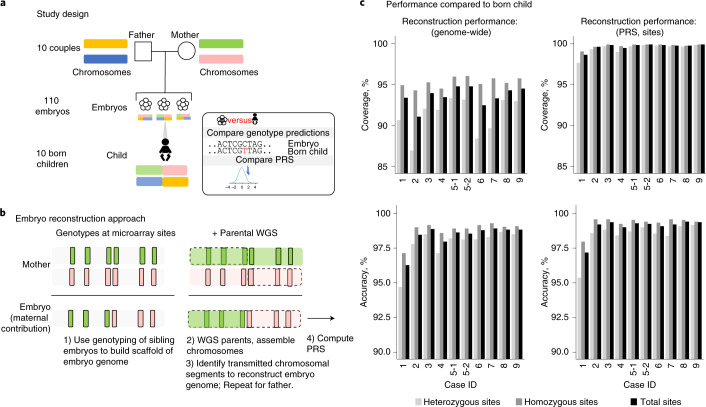
图 1. WGR 和方法。
a、在这个实验中,遗传病阻断课题组分析了10对夫妇的110个胚胎基因组,并与出生婴儿的基因组序列进行比较。根据出生婴儿样本和10个相应的胚胎基因序列结果分析计算了12个PRS模型,并比较了它们的一致性。b 、 WGR采用未来父母的全基因组测序(WGS)和同胞胚胎的单核苷酸多态性(SNP)微阵列基因分型。每个SNP的等位基因测量结果都根据a中所示的亲本来源单倍型进行颜色编码。分子和统计/基于群体的技术相结合,对父母的染色体进行分期,推断每个胚胎的减数分裂重组位置,并纠正在测试单细胞或少细胞胚胎活检过程中引入的错误(方法)。重建的胚胎全基因组用于通过计算PRS并推断对疾病风险影响较大的罕见变异的遗传来预测常见疾病风险。c、通过将WGR基因型与出生婴儿的DNA进行比较,结果显示,在用于多基因预测的位点上,第5天胚胎的基因型准确率为99.0%至99.4%,在第3天胚胎的基因型准确率为97.2%至99.1%。案例1仅包含第3天胚胎,案例2同时包含第3天和第5天胚胎。所有其他案例仅包含第5天胚胎。统计数据根据出生婴儿的基因型(杂合子或纯合子)细分。
结果
本研究中使用的样本来自之前接受过体外受精 (IVF) 且在机构审查委员会 (IRB) 方案下进行过临床 PGT(扩展数据图1 )的夫妇(方法)。阻断遗传课题组从 10 名已有 PGT 结果的出生儿童身上获取了 DNA。非整倍体临床 PGT(PGT-A)显示,110 个胚胎中有 68 个为整倍体,110 个胚胎中有 42 个有一个或多个非整倍体染色体(扩展数据图1和扩展数据图2)。阻断遗传课题组通过对父母双方进行高覆盖率基因组测序和对同胞胚胎进行阵列测量,实现了胚胎的 WGR(方法和补充说明1)。阻断遗传课题组结合使用分子和统计学方法,将亲本变异链接到与单个整倍体染色体相对应的“单倍型”中,确定每个胚胎的减数分裂重组位点,并组装每个重组位点之间的相关单倍型片段,以近似整个遗传的胚胎基因组1。
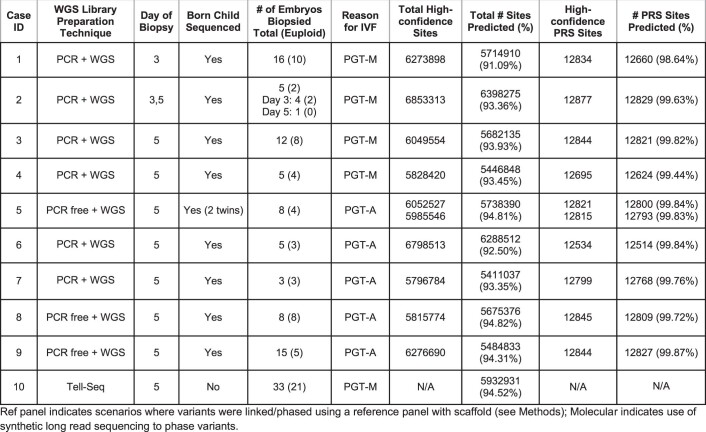
扩展数据图 1. 进行的 PGT 检测的摘要。
共计29名个体在多种测序平台上进行了测序。胚胎活检样本的PGT检测由使用HumanCytoSNP-12 BeadChip芯片进行,样本量从3到33个胚胎不等。在父母和出生婴儿中,对高置信度基因型检出的基因组位点的覆盖率和准确性进行了评估。
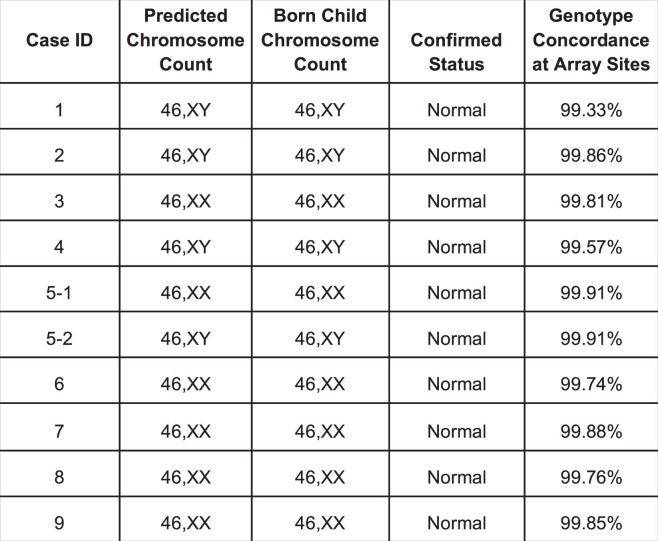
扩展数据图 2. 预测胚胎的非整倍体和基因型并与出生的孩子进行比较。
将每个胚胎染色体微阵列位置的基因型预测结果与出生婴儿全基因组测序 (WGS) 测得的基因型进行比较。预测结果基于 SNP 阵列和父母支持数据。
阻断遗传课题组通过将基因型预测与出生儿童的实际基因型进行比较,评估了WGR的准确性。阻断遗传课题组预测的胚胎中的基因位点平均为580万个位点,位点数量从540万到640万个不等(图1、扩展数据图1和图2)。与出生儿童相比,全基因组预测准确率范围从第3天胚胎活检的96.3%-98.4%到第5天胚胎活检的98.0%-98.9%。阻断遗传课题组推测,与处理和检测第5天滋养外胚层活检中的多个细胞相比,处理和检测第3天卵裂球活检中的单个细胞的性能会有所降低。在四对夫妇中,阻断遗传课题组还使用长片段 DNA(合成长读测序(转座酶联长读测序 (TELL-Seq);扩展数据图1和扩展数据图3)进行了改进形式的文库制备,以捕获罕见变异的阶段(定义为等位基因频率低于 0.1%),并增加了每个家庭中预测的罕见变异的数量和准确性(扩展数据图3)。
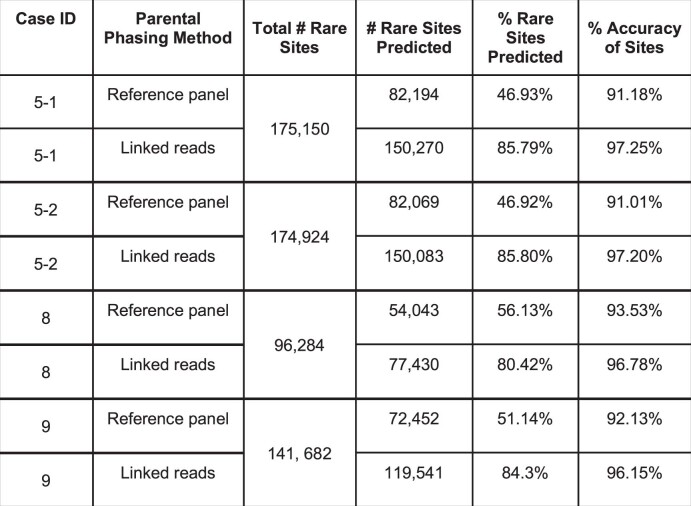
扩展数据图 3. 使用合成长读测序作为分阶段父母的方法,对个别罕见变异进行全基因组重建。
该方法已用于4例病例(病例ID 5、8、9和10)的亲本基因组。由于没有已出生的儿童可供比较,因此无法评估病例ID 10的准确度。等位基因频率<0.1%或未在gnomAD数据库中找到的变异被视为罕见变异。仅评估了Tell-Seq和无PCR全基因组测序方案中高可信度的变异(详见方法部分)。
阻断遗传课题组的方法能够预测胚胎基因组中的罕见和常见变异。为了探索结合这些变异对预测常见疾病风险的影响,阻断遗传课题组使用重建的胚胎基因组来计算 PRS。对于每个胚胎,阻断遗传课题组计算了一组已发表的多基因模型的风险评分,这些模型阻断遗传课题组在英国生物样本库 (UKB) 中基于特定人群进行了验证和校准(方法、扩展数据图4和补充表2)。使用从出生儿童 WGS 中调用的高置信度位置作为“真相”,阻断遗传课题组观察到在来自第 5 天滋养外胚层活检的病例中与多基因风险评分相关的位点的基因型准确率为 99.0–99.4%,在包含第 3 天卵裂球活检的周期中为 97.2–99.1%(扩展数据图1和图1)。阻断遗传课题组对每个胚胎的 PRS 进行了标准化以考虑群体结构,并使用逻辑回归模型将分数转换为预测的疾病几率(方法)。根据胚胎活检计算出的残余 PRS 与根据出生婴儿样本计算出的残余 PRS 之间的相关性为r 2 = 0.947(扩展数据图5)。阻断遗传课题组观察到不同家族的胚胎之间以及家族内同胞胚胎之间的多基因疾病风险存在差异(图2和扩展数据图6)。预测 OR 的最大变化出现在自身免疫性疾病中,这可能是因为 PRS 模型中具有高影响的变异比例较大。虽然白癜风和 1 型糖尿病显示出最高的 OR,但由于疾病相对罕见,这些病例同胞胚胎之间的绝对风险差异不到 10%。绝对风险的最大差异出现在更常见的心脏代谢疾病中。
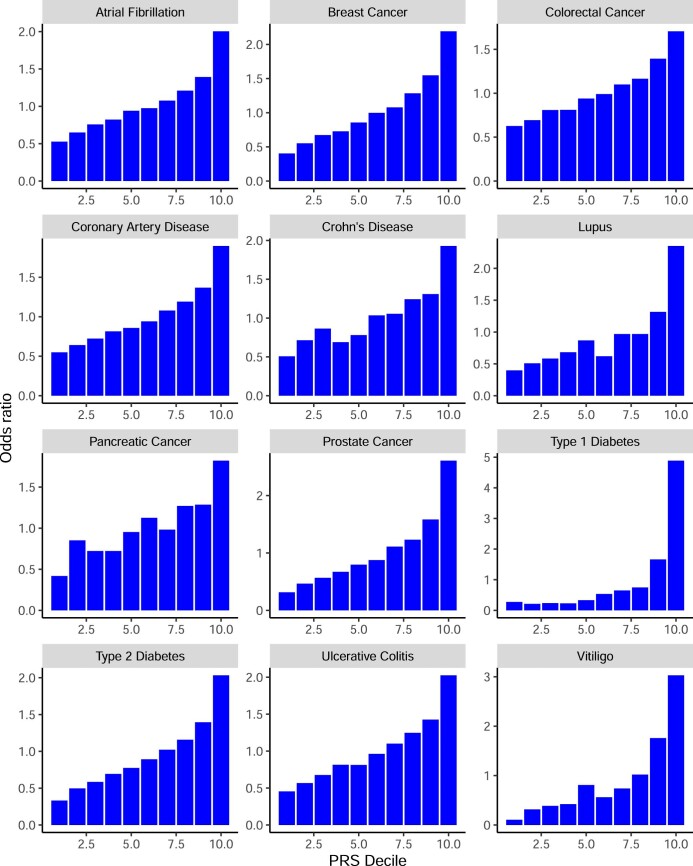
扩展数据图 4. 英国生物银行成年人队列多基因风险评分的表现。
在英国生物银行中计算英国白人个体 PRS 评分每十分位数的经验优势比。
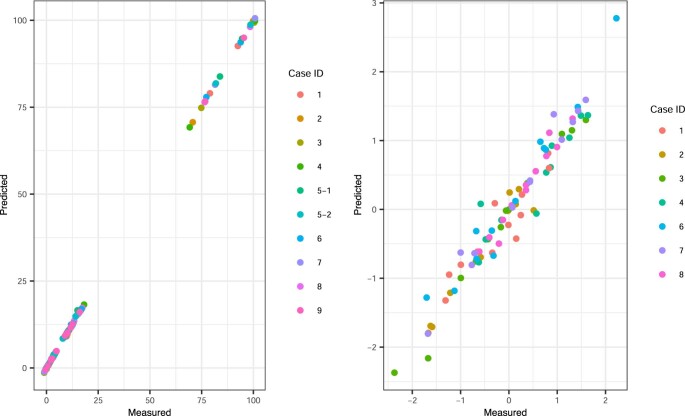
扩展数据图 5. 胚胎预测和出生孩子的多基因风险评分的相关性。
a,说明预测值与测量值(出生儿童)原始多基因风险评分之间的密切相关性,与预测值与测量值之间的基因型一致性一致。b ,预测值与测量值(源自原始多基因风险评分)z值之间的相关性(r 2 =0.947)。病例ID 5和9被排除在本分析之外,因为使用群体血统计算平均中心多基因风险的方法无法解释混合因素。
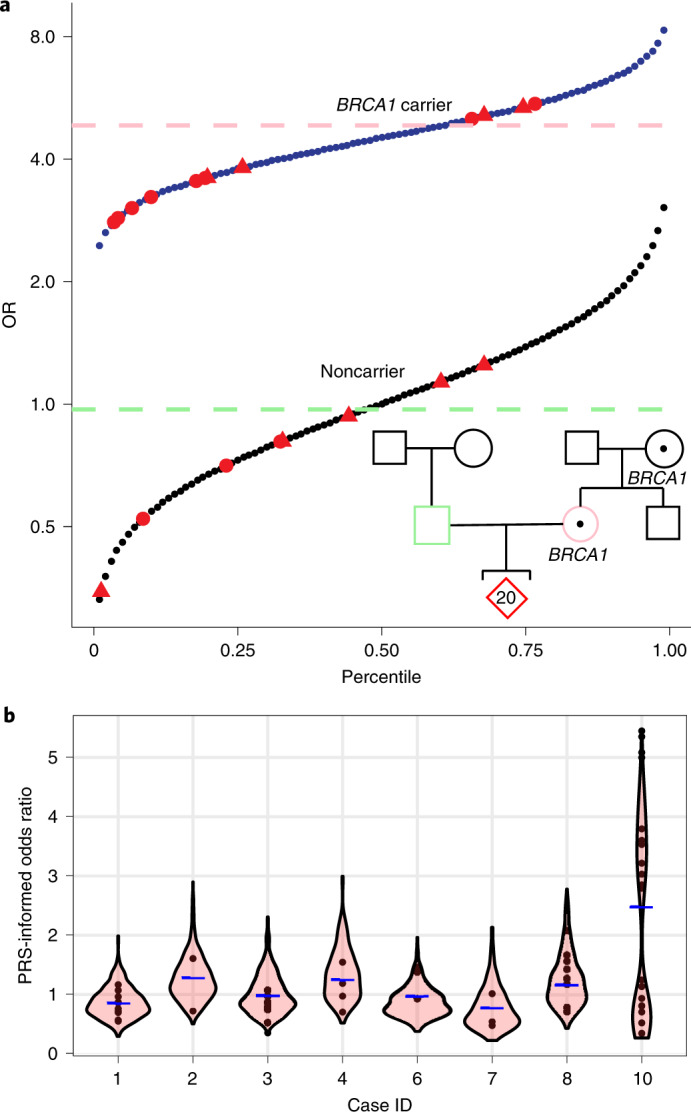
图 2. 家庭内部和家庭间乳腺癌易感性遗传风险的预测差异。
a,对具有乳腺癌家族史、BRCA1变异和多个胚胎(红色)的研究参与者进行多基因/单基因综合预测。13 个胚胎携带致病性BRCA1变异。阻断遗传课题组使用逻辑斯蒂模型对 UKB 中超过 22,000 名具有相关临床和遗传信息的个体进行拟合,以 PRS 和携带者状态作为独立变量,按照 Fahed 等人的方法预测了每个胚胎的患病几率。12蓝线表示BRCA1携带者的 OR 与 PRS 的关系,黑线表示非携带者的 OR。该方法解释了在BRCA1阳性状态下 PRS 效应的降低,这体现在两条线的斜率差异中。女性参与者的 PRS 显示为粉色虚线,男性参与者的 PRS 显示为绿色虚线(投射为女性风险进行比较)。同样,男性胚胎(三角形)也单独显示。b ,显示了十对夫妇及其胚胎的乳腺癌基因组风险。每个胚胎的预测遗传病风险(圆圈)以及500个模拟胚胎结果的小提琴图分布(详见方法部分),其中父母的平均PRS以蓝色虚线表示。致病性BRCA1变异的遗传(a)导致病例10的乳腺癌患病风险变异性增加,并呈现双峰分布。
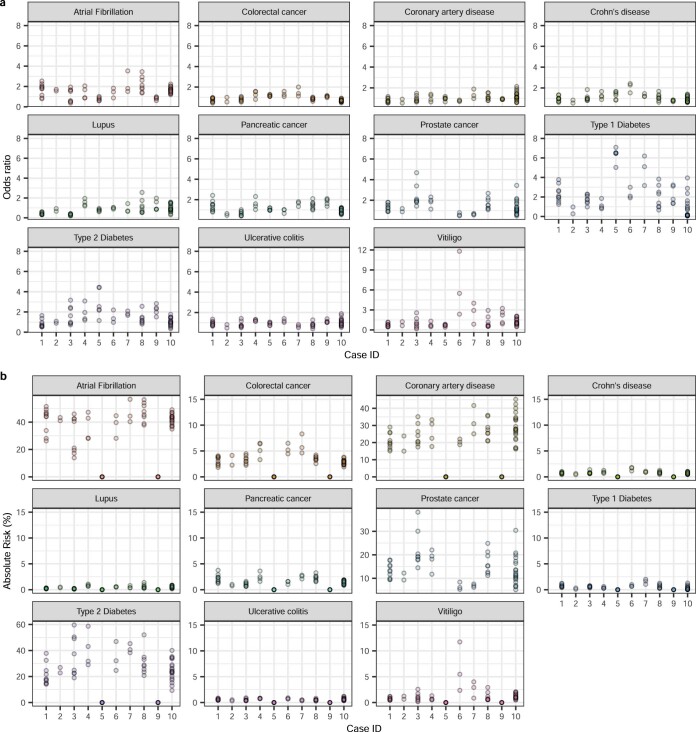
扩展数据图 6. 另外 11 种情况预测疾病风险的变化。
a、预测的疾病相对风险。b 、预测的绝对疾病风险。如前所述,自身免疫性疾病(1 型糖尿病、白癜风)在预测疾病风险方面表现出更大的可变性。注意:携带APC风险等位基因rs1801155 的病例 ID 7 中胚胎的风险完全基于该等位基因。使用竞争风险法(Gail et al 1989)计算心房颤动、冠状动脉疾病、前列腺癌和 2 型糖尿病的绝对风险。年龄和种族特定的总体死亡率和疾病特定死亡率取自 CDC Wonder 数据库(1999-2019 年死亡根本原因)。使用 ELAI 软件确定的非洲/亚洲血统混合比例 >20% 的个体不在本分析考虑范围内。
接下来,阻断遗传课题组研究了在PGT(基因检测)中使用单基因和多基因变异的潜在影响。一对有乳腺癌家族史的夫妇接受了单基因疾病的PGT检测,阻断遗传课题组确认了母亲体内先前发现的致病性BRCA1变异。在该家族的20个整倍体胚胎中,阻断遗传课题组预测其中13个胚胎携带致病性BRCA1变异。在基因组重建后,阻断遗传课题组将携带致病性BRCA1或BRCA2变异的影响与多基因模型的影响相结合,使用Fahed等人12中描述的逻辑回归,计算出每个胚胎的单一风险预测值,并解释了BRCA1阳性状态下PRS效应大小的变化。阻断遗传课题组计算出,携带者中每标准差的PRS OR为1.3,非携带者中每标准差的PRS OR为1.6。不同胚胎的乳腺癌遗传风险预测值相差 15 倍,OR 范围从 0.35(非BRCA1携带者,低 PRS)到 5.35(BRCA1携带者,高 PRS)(图2a)。阻断遗传课题组探讨了单独使用 PRS(目前可用于多基因疾病的 PGT)是否会导致无意中转移高风险胚胎。在这种情况下,在 PRS 低于第 50 个百分位数的 14 个胚胎中,有 9 个携带致病性BRCA1变异。接下来,阻断遗传课题组比较了剩余夫妇的胚胎的乳腺癌风险预测值。除了那些具有BRCA1突变的胚胎外,预测大多数胚胎的乳腺癌 OR 值不到两倍(图2b ),这表明在致病变异对疾病风险影响较大的情况下,乳腺癌 PGT 最有益5、6 。一对未接受单基因疾病PGT的夫妇偶然发现,他们携带APC风险等位基因(rs1801155),该基因与结直肠癌风险增加两倍相关。对这对夫妇的三个整倍体胚胎进行基因组重建后,预测其中一个胚胎携带APC的风险等位基因(扩展数据图6 )。阻断遗传课题组在这些已确定的结肠癌( APC)和乳腺癌(BRCA1)中发现的两种罕见变异,) 基因很可能因估算方法而遗漏,具体取决于所使用的参考人群。由于对致病变异一无所知,有家族病史的夫妇可能会无意中仅根据 PRS 就优先植入突变阳性的胚胎。这种情况的发生频率取决于病情和家族史。例如,乳腺癌遗传基因突变估计占病例的 5% 至 10%,在某些人群13和有家族病史的人群中估计比例更高。
讨论
方法
招募参与者、样本收集和测序
WGS、比对和基因分型
胚胎基因分型和PS分析
父母的单倍型定相
为了对每个亲本中的 WGS 衍生变异进行分期,阻断遗传课题组使用了 SHAPEIT4(参考文献11),使用默认参数,以 UK10K Imputation Cohort + 1000 Genomes 第 3 阶段(EGAD00001000776)作为参考面板,以 PS 单倍型作为支架(补充说明1)。这个支架由约 200,000 个分期变异组成,用于锚定使用参考面板执行的分期(扩展数据图7)。每个染色体都独立且并行地处理;之后合并所有染色体。排除多等位基因位点。为了获得参考面板未代表的罕见变异的额外性能,阻断遗传课题组使用了高分子量 DNA 的链接读取测序(补充说明2)。
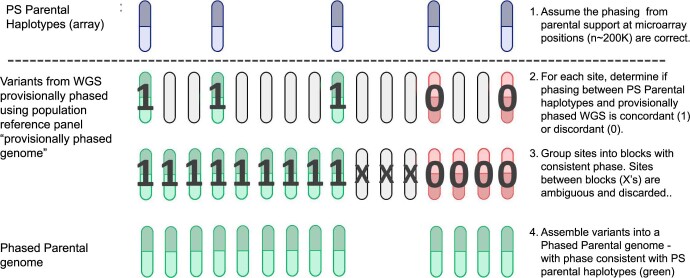
扩展数据图 7. 获取分阶段的亲本基因组。
使用PS亲本单倍型(参见补充说明1)和SHAPEIT4软件的群体参考组对每个亲本的基因组进行分期。PS亲本单倍型作为支架(步骤1),包含约20万个变异。使用群体参考组对父母双方的WGS进行临时分期,并与PS亲本单倍型进行比较。亲本支持单倍型与临时分期WGS之间的重叠位置被标记为具有一致(1)或不一致(0)相位(步骤2),并分组为区块(步骤3)。这些区块之间的间隔区域提示减数分裂重组或亲本一方或双方分期错误;这些位点将被丢弃(步骤3中标记为“X”的位置)。所有剩余位点将用于后续“分期亲本基因组”的组装(步骤4)。
重建方法
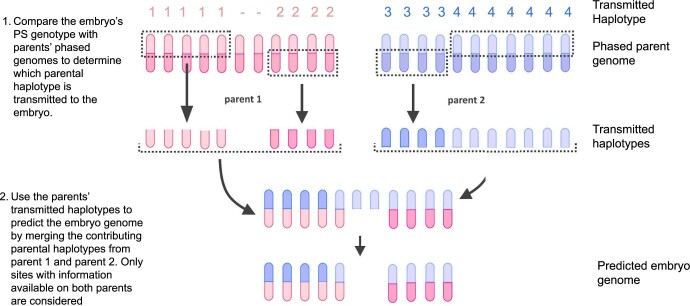
扩展数据图 8.胚胎重建方法。
使用分阶段的亲本基因组和亲本支持 (PS) 胚胎基因型来重建整倍体染色体上的胚胎基因组。分阶段的亲本基因组在扩展数据图7中确定。一位亲本(亲本 1)传递的单倍型显示为粉色(标记为 1 和 2),另一位亲本(亲本 2)分阶段的单倍型显示为紫色(标记为 3 和 4)。在步骤 1 中,胚胎的 PS 基因型决定了哪个亲本单倍型会传递给胚胎(虚线)。胚胎或父母分阶段基因组中缺失的数据被排除在外。在步骤 2 中,将父母双方基因型均有的位置组合起来,以创建预测的胚胎基因组;缺失数据的位置被排除在外。
多基因风险模型
UKB 人口
表型定义
阻断遗传课题组结合 ICD-9 和 ICD-10 代码、自我报告疾病和诊疗程序代码来定义每种感兴趣的表型。所有表型的详细描述见补充表2。
多基因模型
PRS 效应大小
定心和标准化
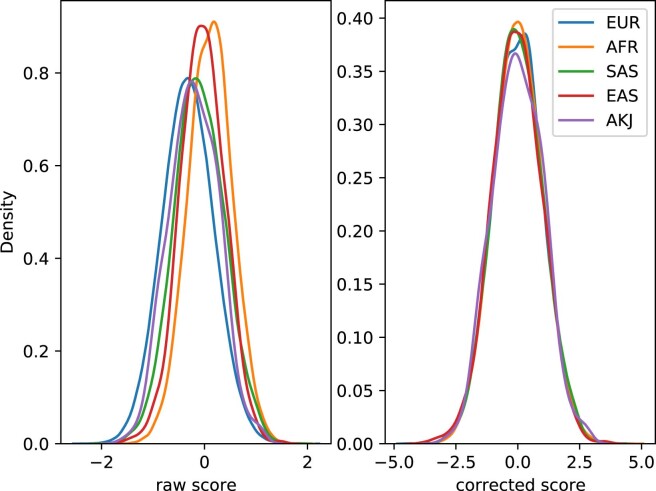
扩展数据图 9. 多基因风险评分的标准化。
英国生物样本库中5个人群的多基因风险评分在中心化和标准化之前(左)和之后(右)。标准化的PRS评分在每个人群中均值约为零,方差为单位。
计算胚胎分数
单基因和多基因风险的整合
PRS的模拟分布
阻断遗传课题组从父母双方的分阶段基因组开始模拟胚胎(连锁方法),在两个母亲或两个父亲的染色体之间添加重组(以近似于配子中的减数分裂重组),并随机组合这些“虚拟配子”。阻断遗传课题组将 PS 单倍型与 WGS 相结合,如上所述,以获得分阶段的亲本基因组。阻断遗传课题组使用 ped-sim(https://github.com/williamslab/ped-sim)与谱系(两个父母和一个孩子)和遗传图谱(https://github.com/cbherer/Bherer_etal_SexualDimorphismRecombination)来模拟重组位点。阻断遗传课题组将从 ped-sim 获得的断点与分阶段的亲本基因组相交以生成虚拟配子,将来自母亲和父亲的虚拟配子组合以生成胚胎基因组,并如上所述计算这些胚胎中的多基因风险。为了生成风险评分分布,阻断遗传课题组对每对夫妇重复此过程 500 次。在另一种方法(非链接方法)中,阻断遗传课题组通过从每个亲本中随机选择一个等位基因来模拟胚胎,并且不对相邻变异是否链接做出任何假设(扩展数据图10)。
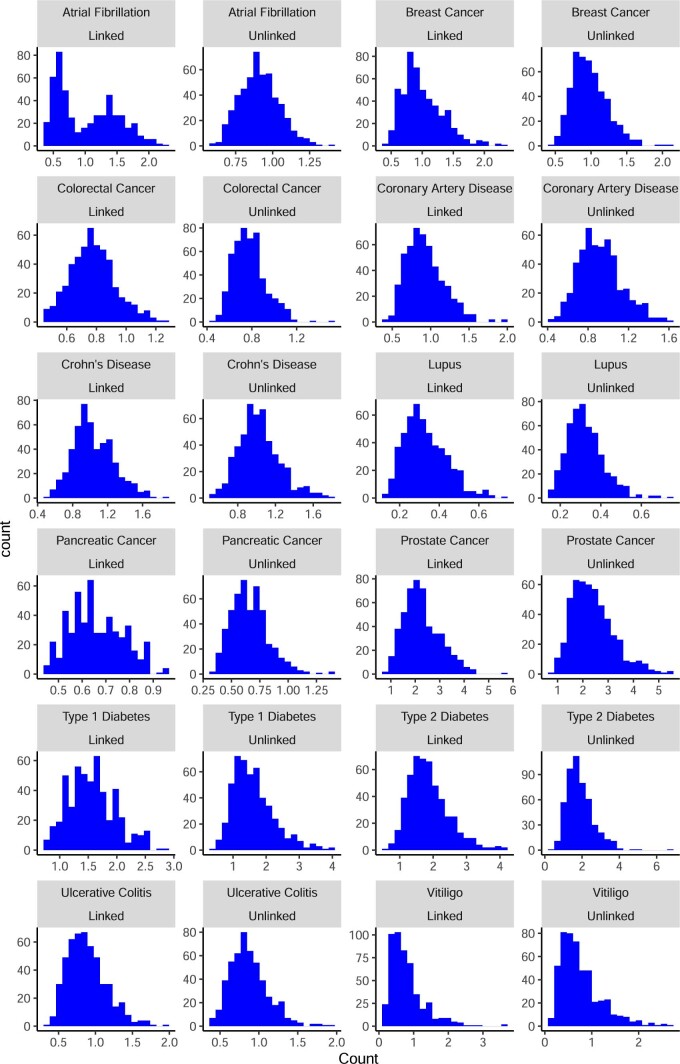
扩展数据图 10. 模拟胚胎风险的分布。
假设一对研究夫妇(病例ID 3)的相邻SNP存在关联和非关联,比较模拟比值比的分布。心房颤动呈现关联SNP的双峰分布,模拟的几个子女预测OR为2,这与基于胚胎的预测结果一致(扩展数据图6 )。关联SNP的模拟采用了《方法》和Caballero等人22中描述的方法。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
