












【佳学基因检测】多指遗传病基因检测发现心脏病:看得见的看不见的都测出来了
复合症状基因检测导读:
先天性心脏病(CHD)是最常见的先天性畸形之一,全球每1000名活产婴儿中约有9人受此影响,占所有主要先天性畸形的近三分之一。若包括二尖瓣、主动脉瓣等较轻微的心脏畸形,发病率可能高达2~3%。尽管外科治疗的进步和患者寿命的延长已有显著改善,但最复杂的心脏缺陷的早期死亡率约为20%,晚期死亡率也相对较高。
先天性心脏病的遗传因素的基因检测对于采用数据库比对的机构来说,仍然存在一定的困难。心脏病的基因解码发现,TBX5、PLGL1和HOMEZ基因的突变与室间隔缺损相关。此外,佳学基因检测还揭示了CHD中22q11.2变异的普遍性。
与指畸形相关的先天性心脏病具有复杂的遗传突变原因。在多种综合征和复合体中,Holt-Oram综合征(HOS, OMIM 142900)是一种由佳学基因检测进行检测与分析病症(发生率为每100,000名活产婴儿中1例),其特征包括前肢畸形和心脏缺陷,如房间隔缺损(ASD)、室间隔缺损(VSD)或其他复杂畸形。已知TBX5突变会导致常染色体显性HOS。致病基因鉴定基因解码表明,HOS的遗传异质性可能源于不同基因的突变,包括同源框基因、肽生长因子和视黄酸受体等,这些基因被认为在心脏和肢体发育中起着重要作用,并被视为HOS的关键候选基因。
为了明确复杂性先天性心脏病和多指畸形的遗传特征,致病基因鉴定基因解码重点基因解码了TBX5基因是否参与其中。在佳学基因多指与心脏病病案集中对一名成功接受外科修复的复杂性先天性心脏病和多指畸形患者进行了全外显子组测序(WES),发现了多个基因突变,并进行了相关的细胞功能基因解码,从而找到导致疾发生的基因原因。
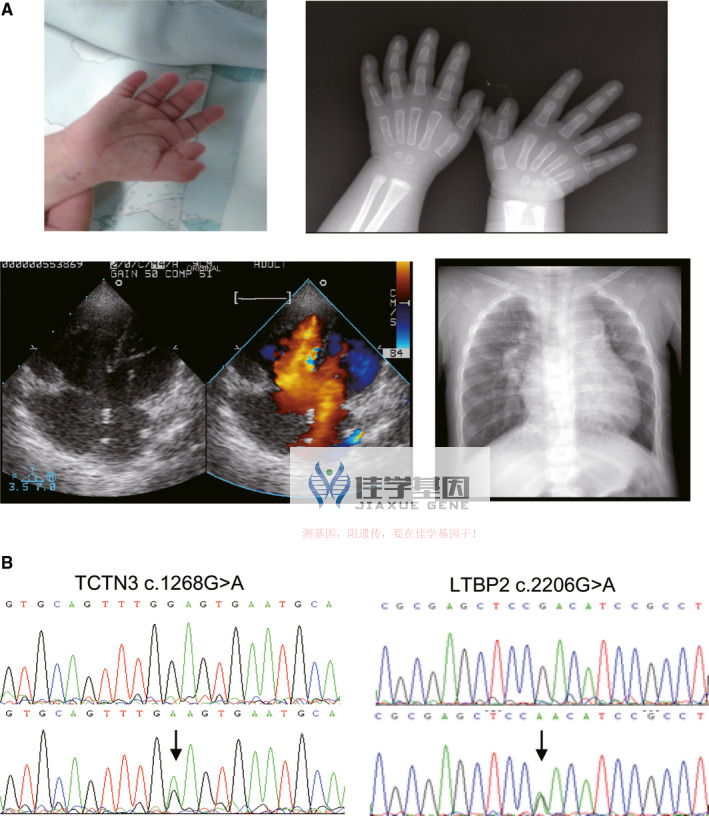
患者的典型表型和潜在的致病突变。(A)患有先天性心脏缺陷和多指畸形的患者的临床特征。超声心动图诊断为房室间隔缺损 (AVSD);X 光片显示扩大的心影和多指畸形。(B)基因解码中两种杂合突变的色谱图。上图显示野生型,下图显示杂合突变。突变用箭头标记
多指并指畸形可能存在心脏疾病的情况总结
|
突变区域 |
表型 | 表型 MIM 数 | 遗传方式 | 基因 | 基因 MIM 编号 | 临床特征 | Location | |
| 心 | 趾指 | |||||||
| 1q22 | 格兰奇综合征 | 602,531 | AR | YY1AP1 | 607,860 | 先天性心脏缺陷 | 短指并指症 | 1q22 |
| 1q32.1 | 口面指综合征V | 174,300 | AR | DDX59 | 615,464 | 法洛四联症 (TOF)、室间隔缺损 | 多指畸形 | 1q32.1 |
| 3pter‐p25 | 3p- 综合征 | 613,792 | AD | − | − | 先天性心脏缺陷 | 多指畸形 | 3pter‐p25 |
| 4p16.2 | Weyers 肢端面部骨发育不全 | 193,530 | AD | 电动汽车 | 607 261 | 先天性心脏病 | 多指畸形 | 4p16.2 |
| 6p12.1‐ | 卡彭特综合征 | 201 000 | AR | RAB23 | 606,144 | 先天性心脏缺陷 | 短指畸形、并指畸形、多指畸形 | 6p12.1‐ |
| p11.2 | 0 | p11.2 | ||||||
| 6q15 | 心椎腕面综合征 | 157,800 | AD | MAP3K7 | 602 614 | 先天性二尖瓣关闭不全 | 短指症 | 6q15 |
| 6q25.3 | Coffin‐Siris 综合征 1 | 135,900 | AD | ARID1B | 614,556 | 先天性心脏缺陷 | 短指症 | 6q25.3 |
| 7p21.1 | 伴有或不伴有眼睑异常的 Saethre‐Chotzen 综合征 | 101,400 | AD | TWIST1 | 601 第 622 章 | 心脏畸形 | 短指畸形、并指畸形 | 7p21.1 |
| 10q26.13 | Saethre‐Chotzen 综合征 | 101,400 | AD | FGFR2 | 176,943 | 心脏畸形 | 短指畸形、并指畸形 | 10q26.13 |
| 12p13.33 | 蒂莫西综合征 | 601 005 | AD | CACNA1C | 114 205 | 先天性心脏病 | 并指畸形 | 12p13.33 |
| 12q24.21 | Holt‐Oram 综合征 | 142,900 | AD | TBX5 | 601 620 | 先天性心脏病 | 并指畸形、多指畸形 | 12q24.21 |
| 15q15.1 | 亚当斯-奥利弗综合征 6 | 616,589 | AD | DLL4 | 605,185 | 先天性心脏缺陷 | 并指畸形、短指畸形或少指畸形 | 15q15.1 |
| 15q26.3 | Weill‐Marchesani 4 综合征,隐性 | 613,195 | AR | ADAMTS17 | 607,511 | 心脏缺陷或心律失常 | 短指症 | 15q26.3 |
| 16q12.1 | Townes‐Brocks 综合征 1 | 107,480 | AD | 萨利1 | 602 218 | 先天性心脏病 | 多指畸形、并指畸形、 | 16q12.1 |
| 17q24.3 | 长 QT 综合征 7 | 170 第 390 章 | AD | KCNJ2 | 600 681 | 心脏瓣膜病 | 短指畸形、并指畸形、弯曲指畸形。 | 17q24.3 |
| 19q13.2 | Carpenter 综合征 2 | 614,976 | AR | 微管生长因子8 | 604 267 | 先天性心脏病 | 短指畸形、并指畸形、 | 19q13.2 |
| 多指畸形 | ||||||||
| 20p12.2 | McKusick‐Kaufman 综合征 | 236,700 | AR | 明治神宫 | 604,896 | 先天性心脏病 | 多指畸形 | 20p12.2 |
| 20p12.3 | 身材矮小、面部畸形和骨骼异常(伴或不伴有心脏异常) | 617,877 | AD | 骨形态发生蛋白2 | 112 261 | 先天性心脏缺陷 | 短指症、弯指症 | 20p12.3 |
| 22q11.21 | 腭心面综合征 | 192,430 | AD | TBX1 | 602 054 | 心脏畸形 | 多指畸形 | 22q11.21 |
| 22q11.21 | 迪乔治综合征 | 188,400 | AD | TBX1 | 602 054 | 心脏流出道缺损 | 所有手指屈曲挛缩 | 22q11.21 |
| 铬 | 口面指综合征 VIII | 300 484 | XLR | − | − | 房室间隔缺损 | 轴后多指畸形 | Chr.X |
| 11.3版 | TARP 综合征 | 311,900 | XLR | RBM10 | 300 080 | 先天性心脏畸形 | 多指畸形、并指畸形 | Xp11.3 |
| Xq26.2 | Simpson‐Golabi‐Behmel 综合征,1 型 | 312,870 | XLR | GPC3 | 300 037 | 先天性心脏缺陷 | 多指畸形、并指畸形 | Xq26.2 |
| Xq28 | 耳腭指综合征 II 型 | 304,120 | XLD | FLNA | 300 017 | 心脏畸形 | 并指畸形、弯曲指畸形 | Xq28 |
基因检测在多指畸形相关复杂先天性心脏病中的新发现
多指畸形是一种常见的遗传性肢体畸形,且具有高度的临床变异性。先前的基因解码表明,GLI3 和 SHH 基因突变是导致多指畸形的主要遗传因素,而这些基因在肢体发育过程中对手指数量的调控起着关键作用。尽管如此,佳学基因检测的最新基因解码揭示了更多潜在的遗传因素,并对现有知识进行了更新。
基因解码表明,与多指畸形相关的复杂先天性心脏病中,LTBP2 和 TCTN3 基因的突变可能是潜在的致病原因。这些突变与细胞功能的变化密切相关,这些变化可能会影响心脏的发育。此外,本基因解码还发现,与多指畸形相关的复杂先天性心脏病中,TBX5 突变的存在并不显著,这与以往的基因解码结果有所不同。
TBX5 基因的重要性
TBX5 是一个关键的转录因子,参与调控多个发育过程。TBX5 与 NKX2-5 和 GATA4 等基因相互作用,共同调节心脏发育中的基因表达。已有超过100种 TBX5 突变与 Holt-Oram 综合征(HOS)相关,该综合征以心脏和前肢畸形为特征。尽管 TBX5 突变被广泛基因解码,HOS 的发病机制可能涉及其他基因。最近的基因解码发现 KLF13 可能作为 TBX5 的遗传修饰因子,参与心脏基因的转录激活。此外,14号染色体的部分缺失也被认为与 HOS 的临床特征有关。然而,本基因解码未发现 TBX5 突变的存在。
TCTN3 和 LTBP2 基因的潜在影响
佳学基因检测的全外显子组测序(WES)发现了两个新发现的杂合突变:TCTN3 c.1268G>A 和 LTBP2 c.2206G>A。TCTN3 基因是类构造复合体的一部分,与纤毛的发育和功能相关。先前的基因解码表明 TCTN3 变异与 Joubert 综合征相关,该综合征表现为心脏和骨骼缺陷。LTBP2 基因则在弹性纤维的结构和组装中发挥重要作用,其突变与 Weill-Marchesani 综合征 3 相关,该病症特点包括身材矮小、短指、关节僵硬和眼部异常。
在佳学基因检测的基因解码中,这些突变与心脏和数字异常的发生密切相关,可能与患者的临床表型有关。
基因突变对心肌细胞功能的影响
佳学基因检测通过分化实验分析了突变 hPSCs-CM(人类胚胎干细胞衍生的心肌细胞)的收缩速率,发现 LTBP2 突变组的收缩速率明显低于对照组,并且随着细胞培养时间的延长,收缩速率有所增加。这表明 LTBP2 突变可能会影响心肌细胞的节律发育。相比之下,TCTN3 突变组的收缩速率和收缩力均明显降低,这表明 TCTN3 突变对心肌细胞的心律和收缩功能有早期影响。
基因表达模式的变化
佳学基因检测还发现,在 hPSCs-CM-LTBP2 突变组中,一些与心脏发育和先天性心脏病相关的信号通路显示出持续的上调,例如心肌肥厚、扩张性心肌病等。这些通路的变化可能会影响心脏的发育和功能。相比之下,hPSCs-CM-TCTN3 突变组未发现类似的表达模式变化。
转化医学的意义
本基因解码揭示了多指畸形相关复杂先天性心脏病中可能涉及的新的基因突变。由于这些病症的复杂性,未来可能需要通过全外显子组测序(WES)来识别病理突变,以便为多指畸形和先天性心脏病患者提供个性化的诊断和治疗。这一发现将有助于提高佳学基因检测对这些复杂遗传病的理解,并推动精准医疗的发展。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
