












【佳学基因检测】色素失禁症(incontinentia pigmenti)基因检测案例
色素失禁症基因检测
色素失禁症又叫做母斑症(IP,Bloch-Sulzberger综合征),是一种多系统疾病,伴有特殊的皮肤病变,这些病变经历四个阶段演变,偶尔也会涉及中枢神经系统、眼睛、头发和牙齿。家族性病例(35%)和散发病例(65%)均由IKBKG基因的致病性突变引起。佳学基因检测从收录的的大量病例中介绍一个非同寻常的病例,该家系中两个患有典型母斑症的同父异母姐妹,分子遗传学分析发现其中一个姐妹携带IKBKG基因中一个可能致病的无义变异,而另一个姐妹却没有携带该变异。强有力的临床判断促使进一步进行分子遗传学基因解码,从而在这个独特的家庭中发现了第二个变异。X染色体失活研究表明,这两个新发生的变异来自父亲。对于那些常见新发生突变的基因,在同一家庭中存在不同的致病突变是有可能的,这对于遗传咨询构成了一个挑战。色素失禁症(IP)是一种由X染色体显性基因致病的皮肤遗传病,是由IKBKG/NEMO基因突变引起的罕见疾病,可影响皮肤、牙齿、眼睛和中枢神经系统。佳学基因报告了母斑症的两个家系病例,并通过基因分析发现了与色素失禁症相关的IKBKG基因中的两种新的突变。另外,不同的基因突变类型可表现出不同的临床表型,而同一基因突变类型也可能显示出不同的临床表型。基因检测通过基因解码提供了进一步研究母斑症基因型和表型的临床病例,并丰富了IKBKG基因的突变谱,为将来色素失禁症的遗传咨询和基因诊断提供了依据。
色素失禁症(incontinentia pigmenti)基因检测案例
关键词:IKBKG新发生的致病变异;X染色体显性遗传疾病;基因诊断;母斑症。
病例介绍
在先进个家庭中,先证者是一名新生女婴,出生后在躯干、四肢和头皮上出现了线性红斑和水疱(图1A、B)。1.5个月时,观察到三种典型的皮肤病变:红斑和水疱、疣状增生和重叠性皮肤色素沉着(图1C)。到5个月大时,病变主要表现为线性色素沉着,没有红斑、水疱或疣状增生病变(图1D)。当先证者4岁时,仍存在一些临床表现,包括双下肢小的线性色素沉着和色素减退、头部局部脱发、牙齿发育不良、左眼视神经萎缩和近视(图1E-H)。2022年9月,先证者1.5个月大的妹妹入院,出现与先证者类似的皮肤病变(图2A、B)。然而,到10个月大时,她手指上仍有明显的疣状增生病变,躯干上反复出现红斑和水疱(图2C、D)。她们的母亲出生时也有类似的皮疹,但症状较轻,没有发现皮肤病变或其他与母斑症相关的症状。其他家人都是健康的。在医生建议下,患者联系佳学基因进行皮肤病致病基因鉴定基因解码,从先证者、她妹妹和双亲的外周血中提取基因组DNA。进行了全外显子组测序、MLPA和IKBKG基因的Sanger测序。在先证者、她妹妹和母亲中检测到IKBKG基因外显子6的c.832C>T(p.Gln278*)突变,而在父亲中未检测到该突变(图3A-D)。
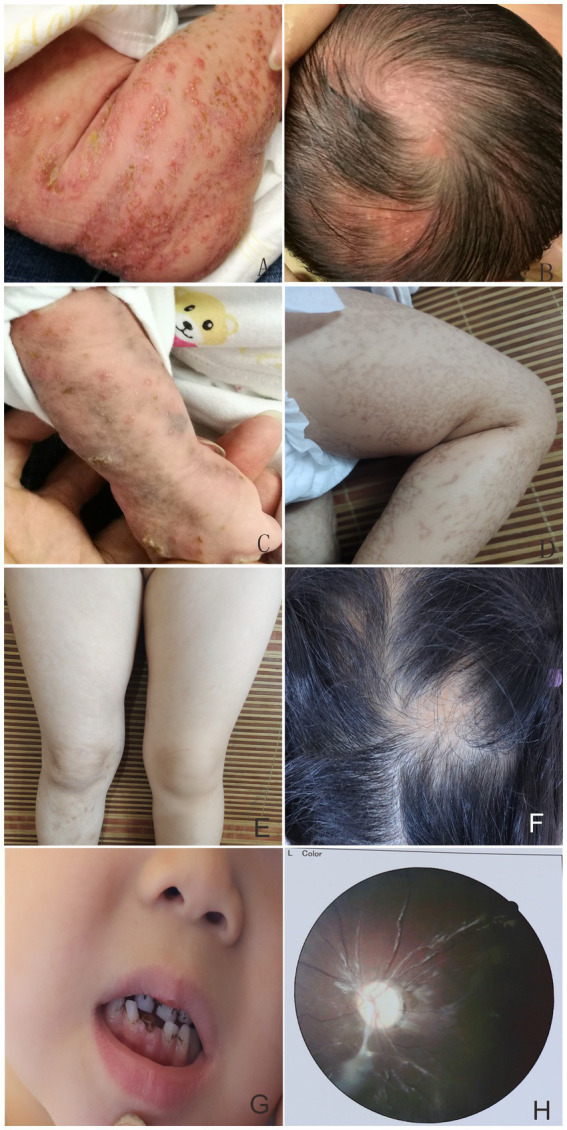
图1: 患儿躯干、四肢及头皮出现线状红斑及水疱(A、B)。1.5月龄时,多个典型皮损与线状红斑及水疱、疣状增生及色素沉着重叠(C)。5月龄时患儿出现色素沉着病变(D)。4岁时,双下肢出现轻微线状色素沉着及色素减退(E),头部局部脱发(F),牙齿发育不全(G),左眼视神经萎缩(H)。
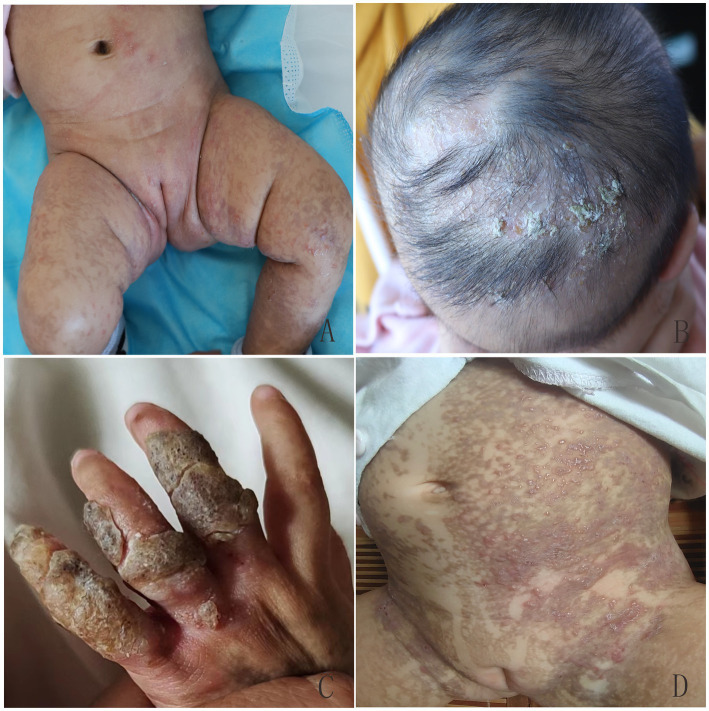
图2:患儿姐姐皮肤出现重叠性红斑和水疱、疣状增生和色素沉着(A),1.5 个月时头皮出现疣状增生和局部脱发(B)。10 个月时手指出现疣状增生(C),躯干出现反复性红斑和水疱(D)。
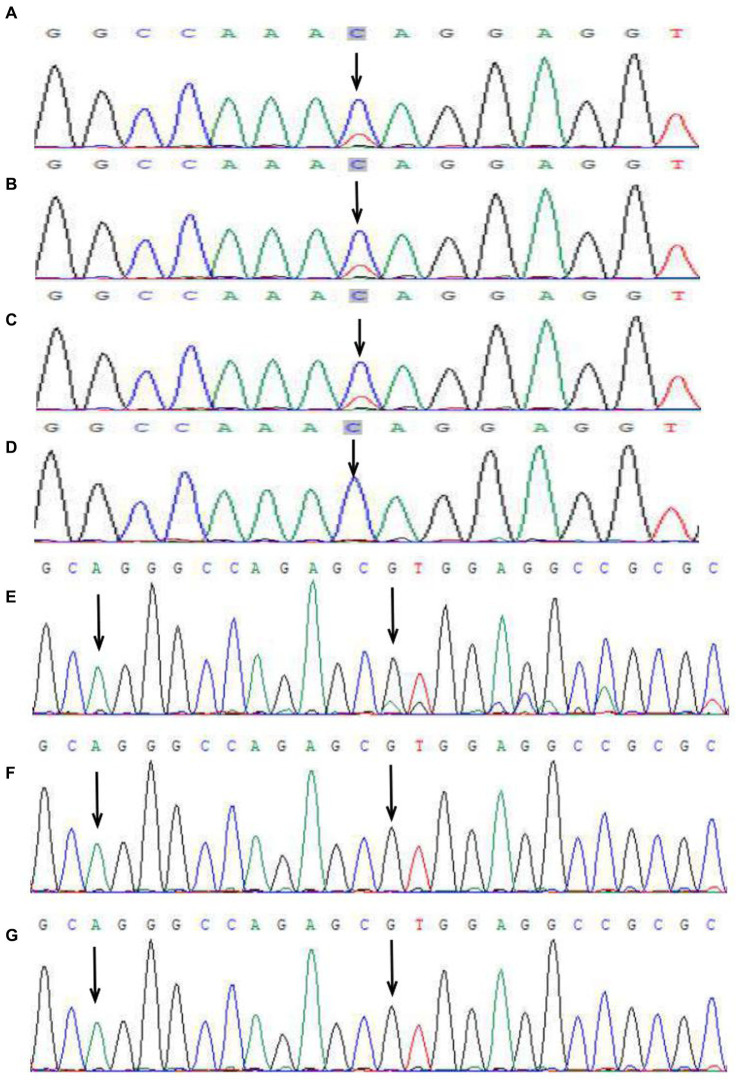
图3:两个家系基因突变测序峰图。先进个家系中,患儿(A)、其姐姐(B)和母亲(C)的IKBKG c.832C>T(黑色箭头),父亲(D)未检测到相同位点的突变(黑色箭头)。第二个家系的患者(先进个箭头)检测到碱基重复突变IKBKG c.614_624dup,显示重复序列(AGGGCCAGAGC)(从第二个箭头开始),产生移码突变,即c.614_624dup(p.Val209Argfs*76)(E)。第二个家系的母亲(F)和祖母(G)均未检测到突变。
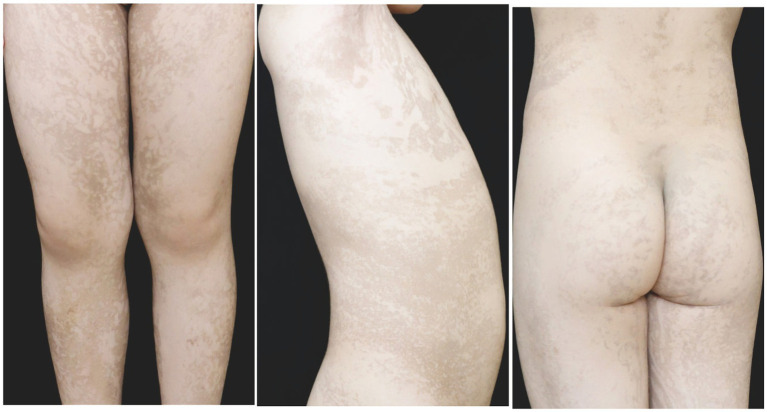
图4:第二个家庭的患者在 4 岁时出现躯干和四肢线性色素沉着。
第二个家系中,患者为一名4岁女孩,躯干及四肢呈线状色素沉着,沿Blaschko线分布(图4)。指甲、头发、牙齿、眼及中枢神经系统均未见异常。出生后躯干及四肢出现红斑和水疱,3个月内皮损逐渐进展为线状色素沉着。患者父母及其他家属均健康。在知情同意后,从外周血提取基因组DNA,对IKBKG基因进行全外显子测序、MLPA及Sanger测序。患者检测出IKBKG基因第5外显子突变c.614_624dup(p.Val209Argfs*76),其母亲及祖母均未检测出相关基因突变(图3E-G)。
色素失禁症(IP) 的诊断基于典型的皮肤损害和 IKBKG 基因突变(3)。未给予特殊治疗。在 4 年的随访期间,皮肤症状持续改善,仅在先证者双下肢观察到少量的线性色素沉着和色素减退。但局部脱发、牙齿发育不全、左眼视神经萎缩和近视仍然存在(图 1E-H)。医生给先证者的先进个家系的姐姐开了外用类固醇药膏来改善手指上明显的疣状增生性病变。1 年后,姐姐的手指上仍存在明显的疣状增生性病变,躯干上反复出现红斑和水疱(图 2C、D)、局部脱发和右眼可疑视网膜病变(图 2B)。在随访期间,家系先进位成员未观察到异常的中枢神经系统表现。第二个家庭中的患者因初次就诊时仅有皮肤色素沉着病变,未接受任何特殊治疗。在 1 年的随访中,家庭成员未观察到指甲、头发、牙齿、眼睛或中枢神经系统的异常表现。
检测过数千名患者后,佳学基因怎么看待色素失禁症?
基因解码基因检测总结到色素失禁症(IP) 是由 IKBKG 突变引起的。基因突变导致该基因编码的功能丧失。贼常见的突变是 IKBKG 基因中外显子 4-10 的缺失。其他突变类型包括错义、移码、无义和剪接位点改变。IKBKG 基因编码 NEMO/IKKgamma,这是一种核因子 κB (NF-κB) 信号转导的调节蛋白。NF-κB 在调节细胞增殖、凋亡和炎症方面起着重要作用。当 IKBKG 未正确表达时,细胞对凋亡信号变得敏感,导致各种炎症反应的产生,这些炎症反应在外胚层细胞中特别活跃。
基因解码还发现,外显子 4-10 的缺失和 IKBKG 基因中的大多数轻微突变都是功能丧失突变,导致女性出现极端 X 染色体偏斜和失活的色素失禁症(IP)表型,男性出现胎儿死亡。 IKBKG c.832C>T(p.Gln278*)和IKBKG c.614_624dup(p.Val209Argfs*76)突变可导致终止密码子过早出现,从而导致IKBKG蛋白的缺失或缺陷,导致NF-κB通路失去正常功能或失调,贼终产生色素失禁症(IP)表型。
关于色素失禁症(IP)反复原因的在教科书上讲的较少。基因解码还发现部分IKBKG缺陷角质形成细胞能逃逸而存活;但残余的IKBKG缺陷角质形成细胞逃脱并存活下来,由于角质形成细胞过度增生和随后的炎症反应的反复,可发生色素失禁症(IP)先进阶段的第二次发作,水泡期通常在4至6个月内消失。但先进家族中先证者的姐姐在近一年的随访中反复出现躯干红斑和水疱,这可能与残留的突变角质形成细胞过度增生和炎症有关。曾经的基因解码基因检测发现,眼部异常往往伴有神经系统表现,并决定了色素失禁症(IP)的严重程度。因此,基因解码基因检测建议密切随访色素失禁症(IP)相关疾病的进展,并采取适当的影像学检查作为预防措施。特别是对于反复的患者,密切随访是必要的。
分子诊断方法很多,贼常见的是全外显子测序、MLPA检测、简单PCR和Sanger测序,不同的检测机构采用的方法不一样,检出率和可信度也有差异。由于IKBKGP1假基因的存在,有时需要多种方法进行基因检测。全外显子组测序未检测到突变,进行MLPA检测和Sanger测序以识别突变。因此,选择合适的基因检测来检测基因突变非常重要。分子诊断有助于诊断色素失禁症(IP),更好地了解IP的基因型-表型相关性,并有助于临床医生指导患者及其家属就预后和未来的生育选择进行咨询。
基因解码基因检测重点在于分析不同基因突变与临床表现的相关性,一些基因检测表明色素失禁症(IP)的基因型和表型之间没有明显的相关性。先进个家庭中的先证者的基因突变类型与第二个家庭中的患者不同,先证者的临床表现比第二个家庭中的患者更严重。然而,这个先证者与她的姐姐和母亲有相同的基因突变,她们的临床表现不同。色素失禁症(IP)的表型异质性归因于X染色体失活的倾斜和对正常X染色体失活的细胞的选择倾向。具体原因正通过基因解码进一步明确,如通过观察和统计大量临床病例的基因型和表型特征。这两项家系研究将有助于进一步研究色素失禁症(IP)表型与基因型之间的相关性。
由于色素失禁症(IP)罕见且表型表达高度多变,它可能是一种没有基因检测无法确诊的全身性疾病,有时会给患者及其家属带来非常高的心理、生理、经济和社会负担。由于没有治好性治疗方法,根据皮肤特征、组织病理学检查和基因检测对IP进行早期诊断对于遗传咨询和及时治疗干预非常重要。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
