












【佳学基因检测】色素失禁基因检测——助力准确诊断
眼科基因检测导读:
色素失禁(或称色素沉着障碍)是一类影响皮肤色素分布和产生的疾病。基因检测在这方面可以提供很大的帮助,因为很多色素失禁的疾病是由特定的基因突变引起的。
佳学基因检测的色素失禁基因检测服务旨在帮助准确诊断这种疾病。以下是一些可能的检测内容和其帮助:
-
基因突变识别:检测可能涉及识别与色素失禁相关的基因突变。例如,常见的与色素失禁相关的基因包括MC1R、SLC45A2等。通过分析这些基因的变异,检测可以帮助确认是否存在特定的遗传标记。
-
准确诊断:基因检测可以帮助医生更准确地诊断病情,避免传统方法中可能存在的误诊或漏诊现象。对于一些症状不明显或者表现多样的色素失禁疾病,基因检测尤为重要。
-
个性化治疗:通过了解具体的基因突变情况,医生可以制定个性化的治疗方案或管理策略,可能包括药物治疗、激光治疗等。
-
遗传咨询:基因检测还可以提供遗传咨询服务,帮助家庭了解遗传风险,并对未来的孩子进行适当的预防或治疗措施。
总体而言,佳学基因检测的色素失禁基因检测通过提供详细的基因信息,有助于更准确地诊断和管理色素失禁疾病。如果你或你的家人有色素失禁相关的症状,考虑进行基因检测可能是一个有效的选择。
目前诊断色素失禁的技术
色素失调症(IP)的分子诊断分析方法应考虑指示病例的性别。这是因为在 色素失调症(IP)女性中,该变异处于组成性杂合状态。这表明它存在于体内所有细胞中。然而,如果合子后突变发生在男性群体中,胚胎嵌合体可使两组遗传上不同的群体共存于同一个体中。此外,表达IKBKG/ NEMO 变异的细胞可能逐渐被消除并最终被清除,这使得男性患者的诊断极其困难。简单的 PCR 检测外显子 4-10 的基因缺失仍然是IKBKG变异筛查的推荐技术,因为复发性缺失占女性 色素失调症(IP)病例的 79% 或全部色素失调症(IP)病例的 70% 。如果未检测到外显子 4-10 缺失,则可使用 Sanger 测序筛查IKBKG编码区和内含子-外显子连接处的点突变和插入缺失,将提高 9% 的诊断灵敏度。除此之外,qPCR 还可用于检测除经典的外显子 4-10 缺失以外的更大的序列变化,这占 色素失调症(IP)病例的约 4%。尽管下一代测序更加广泛且更便宜,但由于假基因IKBKGP1的存在,它被认为无法用于色素失调症(IP)诊断。IKBKG和IKBKGP1都位于 Xq28 区域内,并且有 99% 的序列相同。该假基因的存在使传统的捕获探针数据分析变得困难,因为它减少了读取深度,降低了突变位点定位精度,并导致比对读取不佳,从而导致假阳性结果。然而,下一代测序 (NGS)/全外显子组测序 (WES) 流程分析中掩盖IKBKGP的生物信息学调整利用了该技术,成为检测IKBKG突变的有力工具。凭借此类创新,NGS/WES 无疑加速了IKBKG突变筛查,可替代或补充传统的 Sanger 法。如果使用与女性患者相关的方法,男性患者中发生的低水平嵌合体可能会逃避分子诊断。不应从外周血中提取基因组 DNA,而应使用从疑似表型中选择的组织(即皮肤)进行测试,并分析多种组织,即血液、新鲜皮肤、唾液和精子样本,以检测低水平嵌合体。因此,后者成本更高,并且需要更具体的能力和基础设施。对于基因解码价格敏感的受检者可能以失去敏感性而补偿。
色素失调症的致病基因鉴定基因解码
不同人群中色素失调症的遗传变异
色素失调症(IP)中最常见的基因突变是IKBKG基因中约 11.7 kb 的缺失,该缺失删除了外显子4 至 10。这种突变是 70%–80% 的 色素失调症(IP)患者的发病原因(22-24)。这在欧洲、中国、日本、韩国和印度人群中都有发现。除了11.7 kb的缺失之外,IP还可能由于 IKBKG 基因上的其他类型的突变而产生,包括单核苷酸替换、点突变和小插入/缺失 (indel)。点突变可以是导致蛋白质翻译过早终止的无义突变,也可以是导致氨基酸改变的错义突变。小的 indel 可能导致移码或框内氨基酸缺失。点突变和插入/缺失均可能导致IKBKG mRNA 的异常剪接。这些突变可导致IKBKG蛋白缺失或缺陷,从而产生 色素失调症(IP)的临床症状。基因解码通过分析不同的突变特征,而可以检测出更多的致病性突变,从而提高检出率,降低假阴性结果。除涉及外显子的突变外,基因检测数据库中还收录了中国人群中的涉及内含子 8 的单核苷酸多态性。尽管收录的病例较少,但这种多态性也在高加索人群中有所报道。虽然大多数关于 色素失调症(IP)病例的报道来自西方人群和某些东亚地区,但在非洲、印度、马来西亚和巴西等其他人群中也观察到了 色素失调症(IP)病例。
IKBKG色素失禁的病理生理学
IκB 激酶 (IKK) 蛋白复合物由催化亚基 IKKα 和 IKKβ 以及 IKKγ (NEMO) 组成。IKBKG基因负责编码 IKKγ (NEMO),而 IKKγ (NEMO) 是抑制剂 κB (IκB) 激酶 (IKK) 复合物的调节亚基,而该复合物对于许多基本生理功能所需的 NF-κB 通路激活至关重要。IKK 复合物激活后,IκB 蛋白磷酸化、泛素化和降解会导致激活 NF-κB 复合物的抑制剂被去除。IκB 的缺失使得 NF-κB 能够转位到细胞核中,在那里可以进行目标基因的转录。据报道,活化的 NF-κB 可执行免疫和炎症反应,并参与防止信号蛋白诱导的细胞凋亡。因此, IKBKG基因的功能丧失或缺失会导致IKK功能障碍,从而导致 NF-κB 活性终止。如果没有NF - κB ,色素失调症(IP)细胞对促凋亡信号高度敏感。
在男性嵌合体和女性X染色体异常的情况下,邻近的未发生IKBKG基因突变、表达IKKγ(NEMO)蛋白的角质形成细胞,在接收到正在凋亡或坏死的IKBKG缺陷型角质形成细胞的激活信号后,可发生NF-κB激活。凋亡或坏死细胞产生的激活信号包括危险相关分子模式(DAMP)以及“找到我”信号,如溶血磷脂酰胆碱(LCP)、鞘氨醇1-磷酸(S1P)、核苷酸ATP/AUP和肿瘤生长因子(TGFβ)等。附近表达IKBKG 的角质形成细胞中 NF-κB 的激活将导致趋化因子的产生,例如调节活化、正常 T 细胞表达和分泌 (RANTES)、单核细胞趋化蛋白 (MCP-1) 和嗜酸性粒细胞趋化因子,从而募集嗜酸性粒细胞。此外,还会产生促炎细胞因子,例如 IL-1、TNF-α、IFN-γ、淋巴细胞趋化因子。基因解码发现,IL-1 和 TNF-α 可以上调嗜酸性粒细胞趋化因子的产生,从而吸引嗜酸性粒细胞迁移。募集的嗜酸性粒细胞将发生脱颗粒并释放蛋白酶,导致表皮和身体其他部位发炎 (图1)。
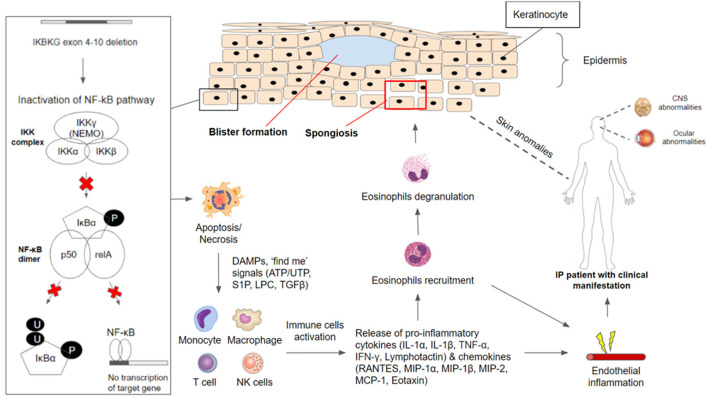
图1:色素失调症中的IKBKG /NF-κB 病理生理学
IKBKG基因是激活核因子 κB (NF-κB) 信号通路所必需的。在无刺激条件下,NF-κB 通过与 NEMO/IKKgamma(由IKBKG编码)结合在细胞质中保持非活性。IKK 复合物对抑制性 NF-κB (IκB) 蛋白的磷酸化导致其蛋白酶体降解并随后释放 NF-κB 二聚体(由 p50 和 relA 亚基组成)。大多数患有色素失调症的个体携带IKBKG基因的常见致病变异,外显子 4-10 缺失,导致 NF-κB 信号通路失活。缺乏IKBKG 的角质形成细胞由于失去对细胞死亡的保护而易发生凋亡/坏死。 DAMPS 和“找到我”信号(ATP/UTP、S1P、LPC、TGFβ)被释放,并作为刺激免疫炎症反应的激活信号。单核细胞、巨噬细胞、T 细胞和 NK 细胞已被证明会释放细胞因子(IL-1α、IL-1β、TNF-α、IFN-γ、淋巴细胞趋化因子)和趋化因子(RANTES、MIP-1α、MIP-1β、MIP-2、MCP-1、嗜酸性粒细胞趋化因子),从而导致嗜酸性粒细胞的募集。募集的嗜酸性粒细胞脱颗粒,释放蛋白酶,帮助降解角质形成细胞之间的粘连。这会导致海绵状水肿和水疱形成,这在色素失调症(IP)患者的第一阶段临床表现中经常观察到。除了主要表现为皮肤病外,色素失调症(IP)患者还经常报告出现中枢神经系统和眼部异常。 NF-κB缺陷的内皮细胞和全身其他细胞趋化因子过度表达,导致嗜酸性粒细胞增多,引发广泛炎症,内皮炎症导致血管阻塞和缺血,导致视网膜和神经系统的表现。
在表皮中,蛋白酶降解张力丝和桥粒,导致细胞内水肿(海绵状水肿)并最终起水泡,这在 色素失调症(IP)的第一阶段可见。随着IKBKG缺陷角质形成细胞的减少(由于细胞凋亡增加、逐渐被表达IKBKG的角质形成细胞所取代以及炎症消退),皮肤病变逐渐消失。此外,TNF 和其他细胞因子可能在炎症早期在表皮中产生,并可能在直接消除IKBKG缺陷角质形成细胞的过程中发挥作用。然而,成功逃脱并在消除过程中存活下来的残留IKBKG缺陷角质形成细胞可能会因角质形成细胞过度增殖和随后的炎症反应而经历 色素失调症(IP)第一阶段的第二次发作。
如果 NF-κB 缺陷的内皮细胞和全身其他细胞过度表达嗜酸性粒细胞特有的趋化因子(如嗜酸性粒细胞趋化因子),则可能导致全身性嗜酸性粒细胞增多症。嗜酸性粒细胞与其他炎症因子相结合会导致广泛炎症。内皮炎症会导致血管阻塞和缺血,导致视网膜和神经系统表现。视网膜动脉阻塞导致区域无血管和灌注不足,从而引发缺血。新生血管形成是其后遗症。在中枢神经系统,脑萎缩和其他神经系统后遗症被认为在视网膜缺血事件中具有相似的血管闭塞性缺血病理生理学。
NF-κB在保护脑内皮细胞和血脑屏障的完整性方面发挥着作用。NF-κB缺陷使内皮细胞易受各种潜在刺激,包括感染。这些刺激上调促炎细胞因子,如IL-6、-8和-10,导致内皮炎症和随后的动脉病变。这解释了全身抗炎在治疗IP患者神经系统表现中的作用。然而,中枢神经系统病变的具体发病机制仍存在争议。
色素失禁的基因型-表型相关性
关于基因型-表型相关性的研究很少。一项对 10 名日本患者及其 3 名母亲的研究表明,有或没有IKBKG基因重排的患者在皮肤外表现方面没有明显差异。佳学基因解码对 42 名 色素失调症(IP)患者进行的研究发现,具有阳性IKBKG致病变异的患者与未具有该致病变异的患者相比似乎具有不同的临床变异。研究观察到,具有阳性IKBKG突变的患者毛发(50 vs. 14%)、牙齿(70 vs. 21%)、眼部异常(45 vs. 29%)的频率较高,中枢神经系统异常的频率较低(20 vs. 35%)。这一差异表明需要深入评估这些组之间的关键表型和基因分型差异。既往研究发现,色素失调症(IP)的临床表型变化很大,从轻度皮肤改变(轻度 IP)到中风和功能性中枢神经系统异常(重度 IP)。致病基因鉴定基因解码发现重度中枢神经系统异常有随机的 X 失活,而无或轻度中枢神经系统异常则有偏斜的失活。另一方面,发现突变类型(常见缺失与点突变)与疾病严重程度无关。这可能是真的,因为 NEMO/IKKgamma 蛋白在调节各种基因表达的复杂信号通路中发挥作用,它的突变会产生不同的表型结果,这可能解释了 色素失调症(IP)中观察到的整个异常谱。
色素失禁症致病基因鉴定基因解码使用的表型评分系统研究色素失调症(IP)患者突变类型与临床表现之间的相关性,结果表明外显子 4-10 IKBKG缺失患者的表型评分变化很大,而次等位基因突变可能具有更广泛的表型后果,因为在 X 染色体失活过程后不久,该突变仍部分活跃。因此,保留一些活性的突变表现出非典型表型,其特征是与经典色素失调症(IP)表型相比,涉及的组织更多。这在具有更严重中枢神经系统和眼部缺陷的 色素失调症(IP)患者中可见,因为不平衡的 X 染色体失活可能会调节疾病的严重程度 ( 25 )。此外,在不同基因组背景下携带相同IKBKG突变的患者的疾病表达差异可以通过其他遗传因素(例如在许多孟德尔疾病中观察到的修饰基因)来解释。迄今为止,色素失调症(IP)中没有显著的基因型-表型关系。然而,研究表明,突变类型、受影响的功能域、X 失活和基因组背景的组合可能导致 色素失调症(IP)表型的变异性。比较详见表格1。
表1:常见外显子 4-10 缺失与其他缺失之间的皮肤外差异
| (n)、人口、性别 | 结果(阳性结果/总数,%) | 皮肤外表型,n (%) | 结论 | |
| 基因检测阳性(delExon 4-10) | 基因检测阴性(外显子 4-10) | |||
| n = 10 日语,女 | 5 / 10, 50% | 脱发,3/5 (60) | 脱发——无 | 他们的研究和其他人的研究中,皮肤外表现没有明显差异 |
| 眼部表现 3/5 (60) | 眼部表现 1/5 | |||
| 神经系统表现 1/5 (20) | 神经系统表现 1/5 | |||
| CVS 表现 - 无 | CVS 表现——无 | |||
| 描述牙齿和指甲异常 | ||||
| n = 42,男女 | 20/34, 58.8% | 头发−10/20 (50) | 头发−2/14 (14) | IKBKG致病变异阳性与阴性IP队列存在临床差异 |
| 白人,27/42 (64%) | 指甲−2/20 (10) | 指甲−2/14 (14) | ||
| 西班牙裔,1/42 (2%) | 牙齿−14/20 (70) | 牙齿−3/14 (21) | ||
| 亚裔,9/42 (21%) | 腭−0/20 (0) | 腭−1/14 (7) | ||
| 黑人,4/42 (10%) | 眼睛−9/20 (45) | 眼睛−4/14 (29) | ||
| 中枢神经系统−4/20 (20) | 中枢神经系统−5/14 (36) | |||
| n = 25,韩国人,女性 | 20/25, 80% (通用外显子 4-10) | 头发−5/20 | 头发−0/5 | 皮肤外表现或表型评分的发生率无统计学显著差异 |
| 5/25, 20% (基因内序列变异) | 指甲−2/20 牙齿−3/20 | 指甲−0/5 牙齿−0/5 | ||
| 眼睛 | 眼睛−3/5 | |||
| −4/20 | 中枢神经 | |||
| 中枢神经系统−5/20 | 系统−2/5 | |||
M,男性;F,女性;CNS,中枢神经系统。
色素失禁症基因检测如何指导治疗
目前的治疗策略需要多学科专家(包括但不限于皮肤科、神经科、儿科、遗传学家和眼科医生)的参与。治疗方法包括症状控制、康复和预防并发症。
对于皮肤出现严重炎症的疣状病变的患者,可使用局部或全身性类固醇和/或局部钙调磷酸酶抑制剂治疗。据报道,类视黄酸可使疼痛的疣状肿瘤消退。医生不应试图用激光治疗色素沉着,因为这可能会加剧皮肤炎症。应强调光保护,因为紫外线照射会加重皮肤病变。
一旦确诊为 IP,应立即进行眼科检查,因为这可能具有视力保护作用。应使用用于筛查早产儿视网膜病变的方案。如果有外周血管病变的证据,则需要在全身麻醉下进行眼底照相和荧光血管造影检查。氩激光可用于治疗无灌注区,可能需要重复激光光凝。雷珠单抗已被描述用于治疗难治性增生性视网膜病变 ,作为激光光凝失败的辅助治疗。斜视和视网膜脱离可以通过手术修复。普萘洛尔被认为是治疗早产儿视网膜病变的潜在方法。
新生儿早期神经系统表现决定了患者的长期预后和残疾的发生。大多数没有新生儿中枢神经系统异常的患儿通常具有正常的身体和认知发育。因此,在准确的皮肤病学检查后进行详细的早期神经系统检查至关重要。应使用脑电图 (EEG) 和脑部 MRI 检查癫痫发作。新生儿期的两个主要治疗目标包括抗癫痫治疗和消炎药物。抗癫痫药物的选择取决于癫痫发作的症状和患者的年龄,而类固醇是首选的消炎药物。最近,抗 TNF 已被成功使用。基因疗法因其在纠正严重脑血管病变方面的潜力而受到研究 。患有神经系统后遗症的患者应尽早接受包括医生、物理治疗师、语言治疗师和职业治疗师在内的康复团队的管理,以减轻神经认知和骨科并发症。对于那些没有神经系统表现的患者,仍应进行常规随访,以发现新的神经系统、神经认知和/或癫痫学表现。
18 岁以下的儿童应定期进行牙科随访,以了解牙齿表现并保持牙齿功能。可能出现的问题包括多发性牙齿发育不全、冠状形态异常、牙颌面矫正异常以及牙齿萌出延迟或缺失。临时假牙和修复治疗可用于替代脱落的牙齿以及牙齿重新定位和排列。当生长停止时,可以开始确定性的种植修复和正畸康复。可能需要进行多学科评估,包括种植牙专家、牙周病专家以及牙颌面矫正和修复专家。
佳学基因检测下一步工作方向
除了将 PCR 和 Sanger 测序作为 色素失调症(IP)基因检测的金标准方法外,还需要进一步创新和改进具有既定策略的 NGS,以提高 色素失调症(IP)分子诊断的灵敏度和特异性。阳性和阴性IKBKG致病变异群之间的临床差异表明需要深入分析这些群体之间的关键基因型和表型差异。对 色素失调症(IP)基因型-表型相关性的更大程度的了解将支持临床医生指导受影响的个人及其家人就预后和未来的生殖选择进行调查和咨询。最后,临床治疗包括确定可能的早期免疫抑制剂以减少炎症标志物,这可能是减少致残的潜在治疗策略,例如视网膜和脑缺血。这可能有助于预防和最佳管理 色素失调症(IP)的严重并发症。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
