












【佳学基因检测】多指医学基因检测:分子诊断阻遗传
骨科基因检测项目:多指基因检测
多指畸形,又称多指畸形或六指畸形,是最常见的遗传性肢体畸形,其特征是多手指或多脚趾,伴有各种相关的形态表型,属于综合征的一部分(综合征性多指畸形),也可能作为单独事件发生(非综合征性多指畸形)。广义上,非综合征性多指畸形分为三类,即轴前多指畸形(桡骨)、中心多指畸形(轴)和轴后多指畸形(尺骨)。多为常染色体显性遗传,具有可变的外显率,由肢体发育前后模式缺陷引起。在人类中,迄今为止已发现至少 10 个位点和 6 个导致非综合征性多指畸形的基因,包括ZNF141、GLI3、MIPOL1、IQCE、PITX1和GLI1。在《多指医学基因检测:分子诊断阻遗传》中,介绍了多指畸形类型的临床、遗传和分子特征,包括最近发现的非综合征性多指畸形的基因和位点。《多指医学基因检测:分子诊断阻遗传》概述了多指畸形的复杂遗传机制,可能有助于遗传咨询和快速分子诊断。
多指医学基因检测:分子诊断阻遗传关键词:
多指畸形、PPD、PAP、手指异常、肢体缺陷、多余手指/脚趾
疾病临床表征数据库
多指症这一术语“poly 表示许多,dactylos 表示数字”起源于 17 世纪的Kerchring。多指症或多指畸形是指出现多余的手指、脚趾或任何复杂的手指部位重复。16 世纪的 Ambrose Parey将这种情况描述为“多余的手指” 。它是出生时最常见的先天性肢体异常之一,表现为各种形式,包括手指完全重复或不完全重复。据估计,一般人群中多指症的发病率为1.6–10.7/1000,活产儿中多指症的发病率为 0.3–3.6/1000,男性发病率往往是女性的两倍。从表型上看,多指畸形是一种极其异质的畸形,右手受累的倾向高于左手,上肢受累大于下肢,左脚受累大于右脚。多指畸形有综合征型和非综合征型两种。两种最常见的多指畸形类型是轴后多指畸形 (PAP),其特征是第五根手指或脚趾有多余的手指,以及轴前多指畸形 (PPD),其大脚趾或拇指侧有多余的手指。而中轴多指畸形是一种非常由佳学基因检测进行检测与分析数字畸形,涉及第二、第三或第四个数字的重复。《多指医学基因检测:分子诊断阻遗传》扩展了Malik (2014)提出的分类。使用 OMIM 和 PubMed [NCBI] 中的“多指畸形”共获得 435 和 3267 个条目。它们包括综合征性和非综合征性轴后多指畸形、轴前多指畸形和复杂多指畸形。
多指畸形分类
多指分类系统和分类依据已在《人体的基因序列变化及其疾病表征》总结。Temtamy和 McKusick (1978)方案是不同遗传咨询师、放射科医生和临床医生中最广泛使用的分类。在该系统中,多指被分为三种类型,即 PPD、PAP 和复杂类型(图1)。
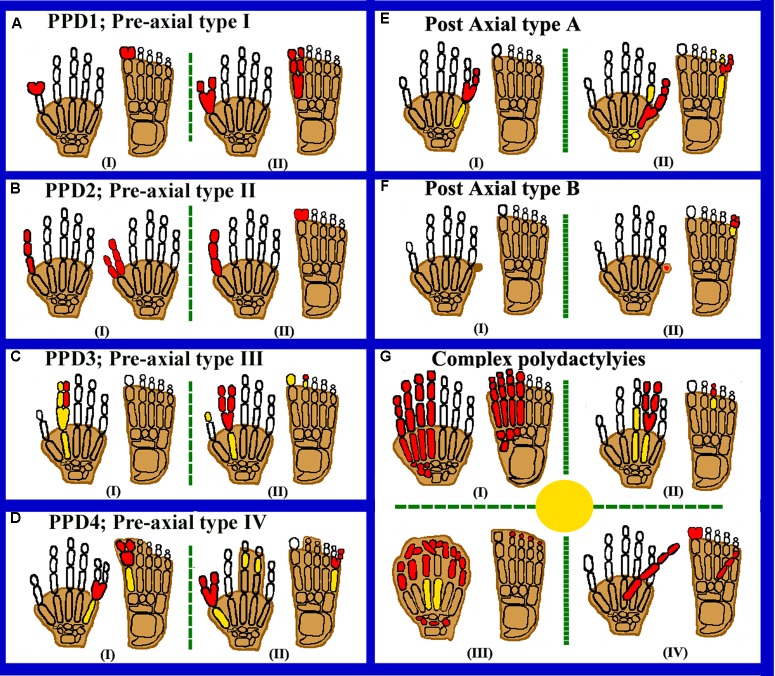
图1:展示轴前、轴后和复杂多指畸形的 autopod 卡通图。红色填充元素表示受影响/多指的手指。黄色填充元素表示发育不良/发育不全的骨骼,阴影手指表示并指。(A)代表 PPD1,包括 (I) 拇指分叉、拇趾多指畸形和 (II) 拇指和拇趾重复。(B)代表 PPD2,包括 (I) 对生三指节拇指和 (II) 非对生三指节拇指。(C)代表 PPD3,包括 (I,II) 第二手指重复。(D)代表 PPD4,包括 (I) 趾蹼(交叉型 I)和 (II) 手指/趾蹼(交叉型 II)。(E)代表 PAPA,具有 (I) 发育良好的第五指和 (II) 第 (5) 第五指更近端的分支。(F)代表 PAPB、(I) 带蒂后小指和 (II) 双裂第五趾带蒂后小指。(G)代表复杂多指畸形 (I),显示镜像前轴重复。(II) 手和脚的中央多指畸形(中轴)。(III) 具有完全并指畸形的 Haas 型多指畸形。(IV) 掌侧/背部多指畸形。
轴前多指畸形 (PPD)
轴前多指畸形是指多指畸形,其附加手指向手的第一指(桡侧;拇指)或脚(内侧)生长。据报道,PPD 的发病率高达 10/300 个新生儿。Wassel 将 PPD 分为七种类型,这对手外科医生非常有用,可帮助他们处理拇指重复。后来, Temtamy 和 McKusick(1978 年)将 PPD 分为四种类型,即拇指/拇趾多指畸形(1 型)、三指节拇指多指畸形(TPT,2 型)、食指多指畸形(3 型)和交叉多指畸形或多指并指畸形(CP,4 型)(图1A–D)。
轴前多指畸形 1 型(拇指多指畸形)
拇指多指畸形是最常见的类型,表现为双指拇指重复(MIM 174400;图图1A)。通常以单侧形式出现,而在双侧病例中,手更容易受到影响,左手 PPD 比右手 PPD 罕见。家族性复发率低,男性患病率高于女性。七指畸形或六指畸形也是一种拇指多指畸形,其中拇指出现三重,从而产生七个手指。家族性 PPD 类型 1 仅显示常染色体显性遗传模式,渗透力降低。 1 型前轴多指畸形是由音猬因子 (SHH) 增强子的序列变异引起的,这种增强子称为极化活性区 (ZPA) 调节序列 ( ZRS )。在肢体发育过程中,前后 (AP) 轴的模式由SHH (MIM 600725) 在称为极化活性区 (ZPA) 的区域中表达决定。一种称为ZRS (MIM 605522) 的顺式调节增强子;可控制 SHH 肢体的表达。ZRS是从人类到鱼类高度保守的近 750–800 bp 的功能元素,位于LMBR1 (MIM 605522) 基因的内含子 5 内。最近,ZRS(进化保守的非编码区)上游 500 bp 的致病变异(称为 pre-ZRS(pZRS))与多指表型有关。
已知拇趾多指症 (MIM 601759) 是一种主要表现或一种独立的疾病类型。拇趾重复的发病率为 2.4/100,000,而南美洲拇指多指症的发病率为 1.65/10,000。在拇趾重复方面,女性比男性受影响较小,大多为单侧,主要影响右脚。迄今为止,非综合征性拇趾多指症的分子基础仍不清楚。
三指节拇指多指畸形(TPT;轴前多指畸形 2 型)
轴前多指畸形 2 型 (MIM 174500),或 TPT (双指拇指置换术),其中拇指有一根额外的中节指骨,其第一掌骨异常长而细,两端都有骨骺(图 1B)。TPT通常是对称和双侧的。TTP是显性遗传,具有不完全外显率。Tsukurov等人首次将2型PPD定位在7q36染色体上。ZRS存在于LMBR1基因内含子5中,该基因是2型PPD的发病机制。
食指多指畸形(轴前多指畸形 3 型)
3 型轴前多指畸形 (MIM 174600) 是一种非常由佳学基因检测进行检测与分析常染色体显性遗传疾病。在这种类型中,食指通常是重复的。一个或两个三指节手指取代了拇指。副指的掌骨显示远端骨骺,由于这种表型,3 型 PPD 与 2 型 PPD 或 TPT 区分开来。“中央多指畸形”和“食指多指畸形”有时被归为一类,被认为是拇指重复变异。额外的手指通常有较大的角度或径向偏差,正常的手指可能不同程度地向尺侧偏移(图1C)。
多指并指症,CP(轴前多指症 4 型)
在多指并指症 (MIM 174700) 中,拇指轻度重复,远端指骨显示径向偏差或拇指宽大且分叉。第三和第四指并指症很少出现。在脚部,第一脚趾显示多指畸形,第一掌骨胫骨偏移且短小。值得注意的是,这种疾病不同于并指畸形,在并指畸形中,蹼内的并指畸形与额外的手指有关。“CP”一词通常用于表示后轴和 PPD 的存在,而主要变化是在手和脚的额外手指轴中观察到的(图 1D)。
在 1 型 CP 中,主要观察到足部 PPD 和手部 PAP。2 型 CP 显示足部 PAP 和手部 PPD 相结合。据报道, ZRS和GLI3基因突变导致 1 型 CP 的出现,该基因也与 PAP A/B 等位基因相关。迄今为止,已报告与不同类型的肢体异常相关的 216 种SHH基因突变。
轴后多指畸形 (PAP)
轴后多指畸形是指第五指上有一个或多个额外的腓骨指或尺骨指(图 1E)。其活产患病率为 1-2/1000,存在一些种族差异。PAP 比 PPD 常见 75%,约 8% 的双侧 PAP 病例(包括下肢和上肢)与许多其他先天性综合征缺陷有关。具体来说,对于 PAP,已确认两种不同的类型,即轴后 A 型,具有多指(完全发育的手指)和轴后 B 型(不完整手指),这两种类型的严重程度、外显率估计和遗传模式均不同。PAP A 型进一步分为八种遗传类型:PAP A1-A7 型和 PAP 8 型(有或没有 Ellis–van Creveld 综合征 (EVC) 表型)。据报道,在人类中,有六种潜在致病基因可导致不同类型的非综合征性多指畸形。这包括 GLI 家族锌指 3 基因 ( GLI3,MIM 165240)、IQ 结构域含蛋白 E ( IQCE;MIM 617631)、锌指蛋白 141 基因 ( ZNF141,MIM 194648)、镜像多指畸形基因 ( MIPOL1,MIM 606850)、成对同源结构域 1 基因 ( PITX1,MIM 602149) 和 GLI 家族锌指 1 基因 ( GLI;165220)。
轴后多指畸形 A 型
在 A 型 PAP 中,完全发育的多余手指与重复的跖骨或掌骨或第五跖骨或掌骨关节相连。重复手指可能有 1 至 3 个骨性元素,具体取决于其大小,这会导致屈曲皱纹和发育良好的指甲。后轴手指和第五趾骨显示出高度可变的关节角度(即 <30–180°)。在大多数情况下,多余手指可能会给日常生活带来问题(无功能性手指)。A 型 PAP 大多为常染色体显性遗传,外显率降低。然而,已发现数个巴基斯坦家庭存在以常染色体隐性遗传方式分离的 PAP,并可进一步分为 8 种类型(图2)。
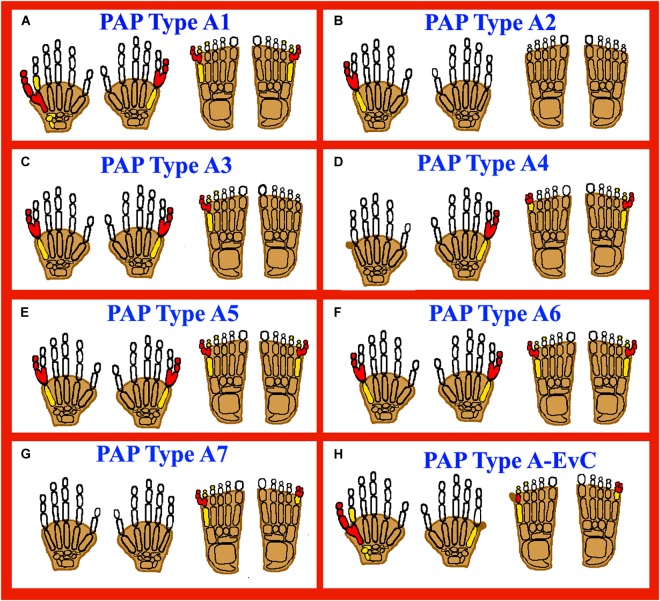
图 2:显示轴后多指畸形 (PAP) 类型 A(PAPA1-PAPA7 和 PAPA 类型 A-EVC)的 autopods 卡通图。红色填充元素表示受影响/多指的手指,黄色填充元素表示发育不良/发育不全的骨骼,阴影手指表示并指畸形。(A)代表 PAPA1,上肢和下肢都有发育良好的多指。(B)代表 PAPA2,多指仅限于上肢。(C) PAPA3,显示上肢和下肢都有发育良好的多指。(D) PAPA4,有发育良好的多指,PAPB 也有报道(皮赘无骨)。(E、F) PAPA5、6:发育良好的多指和脚趾。(G)代表 PAPA7,多指仅限于脚趾。(H)代表 PAP 类型 A-EVC,上肢和下肢均具有 PAP 类型 A 和 PAP 类型 B。
轴后多指畸形 A1 型 (PAPA1)
PAPA1 和 PAPB(MIM 174200)以及 PPD 类型 IV(MIM 174700)以常染色体显性遗传,由位于染色体 7p14.1 上的GLI3基因(MIM 165240)中的致病杂合突变引起。
多指的致病基因鉴定基因解码收录了一个五代家族,其中 15 名患病个体通过全基因组连锁分析具有 PAPA 表型。使用 7p15–q11.23 染色体上的标记 D7S801 获得的最大 LOD 评分为 4.21。因此,在GLI3基因中鉴定出致病杂合变异。同样,基因解码解析了一名手部患有 B 型 PAP 的患者。基因分析鉴定出GLI3基因中致病杂合变异,预计会导致过早终止并可能导致无义介导的 mRNA 衰变。
基因检测机构采用致病基因鉴定基因解码分析了一个来自沙特阿拉伯的三代家庭,其表型包括手部 PAP、宽拇指和手脚并指(皮肤)。遗传和分子分析发现GLI3基因中存在杂合的 2 bp 移码缺失,预计会导致该基因 N 端部分截断(图2A)。迄今为止,已在GLI3基因中发现了 225 个突变,这些突变会导致不同类型的肢体异常,包括多指表型。
轴后多指畸形 A2 型 (PAPA2)
轴后多指畸形 A2 型,具有常染色体显性遗传(MIM 602085),位于 13q21–q32 染色体上,致病基因尚未确定。Akarsu等人(1997 年)对一个表现出 PAPA 特征的家族进行了连锁分析,并排除了该家族的 7p15–q11.23 基因座,该基因座已知会导致 PAPA1(MIM 174200)。该家族与 13q21–q32 染色体有连锁,标记 D13S1230 的最大 LOD 得分为 2.35。
此外,佳学基因等机构利用标准核型分析,在一位患手双侧 PAP 的男孩中,发现了 13 号染色体长臂上存在杂合新生倒置重复。FISH 分析和阵列比较基因组杂交 (aCGH) 分别进一步证实了杂合重复(图2B)。
轴后多指畸形 A3 型 (PAPA3)
具有常染色体显性遗传(MIM 607324)的轴后多指畸形 A3 型位于 19p13.2–p13.1 染色体上。多指采用致病基因鉴定基因解码技术确定了标记 D19S221 的最大 LOD 得分为 5.85,对应的物理距离约为 2.5 Mb。16 名受影响的个体表现出手和/或脚上发育良好且功能齐全的轴后多指等表型。七名受影响的个体患有手部多指畸形,未观察到其他表型(图2C)。
轴后多指畸形 A4 型 (PAPA4)
A4 型轴后性多指畸形最早见于一个有 11 名患者的六代家族。在患者中,PAPA 和并指表型在上肢和下肢均存在差异,没有其他相关异常(图 2D)。全基因组筛查的遗传和分子分析确定标记 D7S1799 的最大 2 点 LOD 得分为 3.18。PAPA4 显示常染色体显性遗传(MIM 608562),位于染色体 7q22.1 上,致病基因尚未确定。
轴后多指畸形 A5 型 (PAPA5)
一个来自偏远地区的巴基斯坦近亲家庭,据称患有常染色体隐性遗传,双手和双脚均表现出 PAP A 型(双侧)的特征。而受影响的个体及其正常父母没有观察到其他异常(图2E)。多指的基因解码使用全基因组连锁分析,通过对高度多态性的微卫星标记进行分型,发现在 13q13.3–q21 染色体上存在最大 LOD 得分为 3.84 的连锁。PAPA5 (MIM 263450) 被定位到 17.87cM 区域 (D13S1288 和 D13S632 标记),并收录了定位到 13q13.3–q21 染色体的常染色体隐性 PAP 的第一个证据。
轴后多指畸形 A6 型 (PAPA6)
多指基因检测发病原因在研究一个巴基斯坦近亲家庭时,发现该家庭双手和双脚均存在发育良好的(双侧)PAP(图2F),在 4p16.3–p16.2 染色体上确定了一个 6.53 Mb 的连锁区间。对 27 个高度多态性的微卫星标记进行了分型,在标记 D4S412 处获得了最大 3.38 的多点 LOD 得分。利用 aCGH 分析和全外显子组测序等技术,多指发发病原因基因检测在ZNF141基因 (MIM 194648)中确定了一个错义突变 (p.Thr474Ile) ,该突变与家族内的疾病表型分离并归类为 PAPA6 (MIM 615226)。
轴后多指畸形 A7 型 (PAPA7)
多指的致病基因鉴定基因检测在IQCE基因 (MIM 617631)中发现了一个纯合剪接位点变异,并使用 WES 和小基因检测在一个巴基斯坦血亲家族中进行了验证,该家族中有 5 名患者。对该家族中两名患病成员进行了外显子组测序,随后进行了桑格测序,以在家族内分离具有疾病表型的变异。PAPA7 (MIM 617642) 的特征是 PAP 仅限于下肢,指甲发育良好(图2G)。IQCE基因有 22 个外显子,编码一个含有 695 个氨基酸的蛋白质,位于染色体 7p22.3 上。迄今为止,在与局限于下肢的 PAP 相关的 IQCE 基因中,仅收录了一种剪接受体位点变异(c.395-1G > A;p.Gly132Valfs ∗ 22)。IQCE 的蛋白质产物与EFCAB7结合形成蛋白质复合物,作为刺猬(Hh)信号的正介质。IQCE-EFCAB7 复合物进一步与 EVC-EVC2 形成的第二个蛋白质模块相互作用,将其束缚在纤毛基部(Pusapati et al., 2014)。EVC或EVC2突变会导致埃利斯-范克里维尔德综合征 (EVC),该病的特征是 Hh 信号减弱,多指畸形是其表型之一。因此,IQCE 中的致病突变被认为会导致与异常 Hh 信号有关的多指畸形表型。
伴有或不伴有 EVC 表型的轴后性多指畸形 A 型 (PAP A 型-EVC)
多指畸形的致病基因鉴定基因检测收录了三个肢体异常家族(两个来自土耳其,一个来自巴基斯坦)中位于 12q13.3 染色体上的GLI1基因(MIM 165220)的纯合功能丧失变异。巴基斯坦家族中的四名受影响个体表现出单独的 PAP 特征(图 2H)。另外两个来自土耳其的家族中有四个患者表现出与身材矮小、房间隔缺损 (ASD)、轻度指甲发育不良或膝外翻相关的 PAP,这些症状被归类为典型的 EVC 综合征特征。有趣的是,所有具有GLI1纯合功能丧失变异的患者都具有共同的 PAP 表型。而来自巴基斯坦的家族则具有单独的 PAP 特征,未观察到其他异常。GLI1 基因(MIM 165220; NM_005269.2 ) 有 12 个外显子,编码 1106 个氨基酸蛋白质,位于染色体 12q13.3 上。迄今为止,GLI1基因中仅收录了三种功能丧失突变(c.2340G > A;p.Trp780、c.1930C > T;p.Gln644 ∗、c.337C > T;p.Arg113 ∗ ) ,导致 PAP 和 EVC 表型(Palencia-Campos et al.,2017)。多指畸形的致病基因鉴定基因解码得出结论,GLI1 患者表型谱的多变性可能与突变的位置和剂量效应有关。GLI 蛋白家族(GLI1、GLI2 和 GLI3)是 Hh 信号通路的转录因子,参与胚胎发育过程中的细胞增殖和模式形成。
轴后多指畸形B型
在 PAP B 型中,多指畸形在各种人群中最为常见,多余的手指可能发育不全,因此以皮肤的形式出现,从可忽略不计的小隆起迹象到第五指尺侧的棘状突起,或一个 2-3 厘米长的结节状“带蒂后小指”,通常带有指甲(图1F)。第五指沿着该小结节的关节位置多变,通常通过一个小的皮桥连接。左手和上肢最易受到影响。这种多指畸形的遗传学更为复杂,因此估计的外显率约为 43%。
《人的基因序列变化与疾病表征》,有几种显性遗传基因位点与 PAP 有关,包括位于 7p14.1 上的 PAP1 和相关基因GLI3(MIM 174200)、位于染色体 13q21–q32 上的 PAP2(MIM 263450)以及也具有 PAP-A/B 特征的 PAPA3,染色体地址为 19p13.1–13.2(MIM 607324)。多指的致病基因鉴定基因解码还收录了位于染色体 7q21–q34 上的具有 PAP-A/B 表型和部分皮肤并指的 PAP 基因位点(MIM 608562)。
复杂类型的多指畸形
复杂多指畸形被单独分类,因为它们具有与 PAP 或 PPD 不同的表型(图1G)
镜像多指畸形 (MIP)
在 MIP(MIM 135750)中,后指重复,而前指完全被后指逆序交换。因此,额外指的排列从中央指开始按降序排列,例如小指、无名指、中指、食指,以及中指、无名指、小指,拇指/拇趾缺失(图1G)。多指畸形的致病基因鉴定基因解码收录了一个手指完全重复的个体,右手有 9 个手指,左手有 10 个手指,因此双侧手指结构复杂。单独的 MIP 非常罕见,为常染色体显性遗传,通常被描述为 Laurin-Sandrow 综合征 (MIM 135750) 的一部分。MIPOL1基因 (MIM 606850) 突变导致位于 14q13 染色体的 MIP 表型之一。据多指畸形国际数据库,PITX1基因 (MIM 602149) 突变也会导致伴有下肢畸形的 MIP 。迄今为止,已收录PITX1基因中有 17 个突变导致不同类型的肢体异常。
中轴或中央多指畸形
中轴多指畸形或中央多指畸形是一种“隐藏的”重复畸形,伴有明显的并指畸形,或手掌中部有并甲畸形,可能以组织块的形式出现,但并非所有的中轴多指畸形类型都是隐藏的(图1G [II])。中央多指畸形,如第二指重复,是显性遗传的。畸形通常是双侧的,第四指重复最常见;这些重复比食指重复更常见。
掌侧和背侧多指畸形
帕尔默多指症是一种非常由佳学基因检测进行检测与分析疾病,多指通常从足趾腹侧或背部长出。多指可能表现为发育不良的指骨或小皮肤(赘生物),或发育良好的手指,有或无指甲,并以钩状植入足趾(图1G [III])。也有基因解码发现,手部有可移动的重复手指,并且手指起源于腹侧或手掌侧。同样,基因解码在2007 年记录了起源于足背的重复手指。
Haas 型多指畸形
在 Haas 型多指并指畸形 (MIM 186200) 中,所有手指均在皮肤上融合,并且蹼中有一根后轴或前轴额外射线。由于完全并指畸形,手指的运动受到限制,相邻手指的融合使手呈杯状 (图1G [IV])。人的基因序列变化与疾病表征一般将 Haas 型多指畸形归类为 5 型并指畸形。根据证据,人们观察到 Haas 型多指畸形在遗传上具有异质性。已知 ZRS和GLI3的突变会导致 Haas 型多指畸形。
多指畸形发生的致病基因突变
目前的遗传和分子分类表明,至少六种不同类型的多指畸形是由两个不同基因突变引起的,即SHH增强子ZRS和GLI3,导致PPD1、PPD2、TPT-PS、PPD4、PAPA1和Haas类型。因此,这两个因素的重要遗传作用以及GLI3和SHH之间密切的SHH信号通路在肢体发育过程中的作用不容忽视。在人类中,在妊娠4至8周之间,肢体胚胎发生过程开始,导致肢芽从侧板中胚层长出。不同类型的信号中心在生长中的肢体中发育,指导前/后模式形成的机制,即决定指趾的形成和形态发生。 Sonic Hedgehog-Patched-Gli (SHH-Ptch-Gli) 通路失调会导致多种人类疾病,包括出生缺陷、骨骼异常和癌症。SHH-Gli3 激活的 Ptch 转录通路元件高度保守,在控制 AP 肢体模式方面非常重要。SHH-Gli3 通路中的主要关键参与者包括 Smo、SHH、Gli3、IQCE、Ptch1 以及对 AP 模式也至关重要的视黄酸和骨形态发生蛋白 (BMP)。
Ptch1 和 Smo 也是 SHH 通路中的中间基因,Smo 在 SHH 存在时激活 SHH 信号。而当没有 SHH 时,Ptch 受体抑制 Smo 功能,从而起抑制作用。此通路中最重要的基因是 GLI3,GLI3的致病突变可导致前轴性多指畸形和后轴性多指畸形表型。Gli3 有两种不同的形式(Gli3R 和 Gli3A)。Gli3R 作为阻遏物存在,在 SHH–Ptch 相互作用发生时被 Smo 转化为激活的 Gli3A。Gli3R/Gli3 的比例直接影响指趾类型和数量的发育。然而,由于多指畸形发病机制中其他几种基因的复杂相互作用以及双功能转录开关 ,因此致病基因鉴定基因解码正在开发出与 Gli3 突变的确切基因型和表型相关性。
这些关键参与者(GLI3 或 ZRS)参与肢体表型的发展及其在信号通路中的作用非常重要。然而,多指畸形的复杂性在于遗传异质性现象。由于 GLI3 基因编码序列或800 bp ZRS区域内的致病序列变异导致多指畸形类型范围很广,因此预计不同种族和人群之间会存在遗传异质性。致病基因鉴定基因解码描述了强调轴前多指发病机制的详细ZRS分子机制。许多与综合征和非综合征多指畸形/肢体畸形病因有关的遗传因素仍有待确定。对新发现的基因进行分类是当务之急,这可能有助于临床医生和研究人员清楚了解分子发病机制并快速进行基因诊断。
关于多指基因的共识性意见
人类遗传学方面的进展揭示了几种新的孤立性和综合征性多指畸形类型,这加深了多指发生的基因原因对负责遗传肢体发病机制的几种基因的认识和理解。多指畸形的遗传学非常复杂,不仅限于孟德尔遗传。遗传和等位基因异质性、表观遗传因素、相关基因、增强子/抑制子的作用以及不同类型的环境和发育因素等机制起着非常重要的作用。大量详细的多指畸形遗传学、流行病学、分子学和胚胎学研究观察到表型、患病率、传播和表现度存在显著差异,并表明表型异质性的病因很高。此外,其他几个因素,包括表现力差异、不完全外显率、遗传背景差异、环境影响和表观遗传现象,可能在人类差异表型的形成中发挥了作用 。过去,许多作者认为需要有组织的概述,试图设计一个多指畸形的分类系统。《人类疾病的临床表征与基因位点》列出了不同的多指畸形分类及其优缺点。
随着RNA测序、系统生物信息学、下一代测序(NGS)等新技术的出现,人们将发现新的基因和致病突变,这可能有助于在不久的将来建立基因型-表型相关性。RNA-Seq可以分析不断变化的转录组、转录后修饰、可变剪接转录本、SNP/致病变异、基因融合和基因表达。近年来,WGS/WES(NGS)极大地加速了家族性和散发性病例的诊断,提供了快速、准确且经济有效的基因筛查。适当的基因型-表型相关性可能有助于未来的基因检测,控制医疗障碍,增进多指发生的基因原因对新发现疾病及其相关基因的了解,从而提供通过NGS技术识别的大量变异的实质性知识,这些知识近年来可能用于筛查和分子诊断。此外,致病基因中特定致病变异的影响可能会导致相关表型,而修饰基因中的不同变异可能解释某些情况下的表型变异。将大规模测序数据与正确的表型信息相关联可能有助于使用最新的成簇调节间隔短回文重复/CRISPR 相关蛋白 9 (CRISPR-Cas9) 技术开发预测表型模型,这可能有助于理解肢体发育途径,有助于阐明多指畸形的发病机制并开展实验性治疗研究。这些策略可能会确定治疗干预的最高产量分子靶点,这仍然是未来研究的挑战。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
