












【佳学基因检测】人的外表中的牙齿异基因检测及其科学依据
基因解码基因检测在揭示综合征性缺牙(TA)的遗传学方面取得了显著进展,最常见的孤立性缺牙的病因对于数据库比对来说仍然困难。基因解码已确定了一些新的基因和变异,揭示了牙齿发育过程中相关基因的新途径。此外,随着新一代测序等新研究方法的应用,基因解码基因检测进一步为缺牙的寡基因遗传模型提供了更多证据,这有助于解释该病的表型变异性。在《人体的基因序列变化与疾病表征》中,佳学基因介绍目前有关人类综合征性和孤立性缺牙的遗传机制,并强调结合新一代测序方法识别致病基因和修饰基因的价值。
关键词:缺牙,病因,基因,遗传
1. 牙齿基因检测的遗传学基础导读
牙齿发育需要口腔上皮与神经嵴衍生的间充质之间的多次信号相互作用,并受到多种信号分子及其下游信号通路的精确控制。这些信号相互作用使得牙齿能够形成具有高度特化结构的不同类型的牙齿,如门牙、犬齿、前磨牙和臼齿。任何发育过程中的干扰都可能影响牙齿的生长、分化与模式形成。
牙齿发育不全(TA)是指由于牙齿发育过程中受到干扰,导致一颗或多颗恒牙先天性缺失。TA 是最常见的牙齿发育异常,并且可能是约150种综合征的标志性表现,虽然它也可独立存在,偶尔发生或家族遗传。TA 的遗传模式包括常染色体显性、常染色体隐性或X连锁,表现出明显的渗透力和表现度差异。根据缺失牙齿数量,TA 被分为牙发育不全(缺失不超过5颗牙齿)或少牙(缺失6颗及以上牙齿)。如果所有恒牙缺失,则称为无牙,通常与TA的综合征形式相关。
TA的发生率存在显著差异,并且在不同性别和种族群体中表现出不同的患病率。研究显示,单颗牙齿缺失的患病率为3%至10%,而少牙的患病率较低,仅为0.1%至0.5%(不包括第三磨牙)。若包括第三磨牙,患病率则可高达25%。最常见的缺失牙齿包括下颌第二恒牙、上颌侧门牙和上颌第二恒牙。女性较男性更易发生TA,比例为3:2。
TA的发病偶尔与外部因素相关,如感染、创伤、化疗或放疗,但大多数情况下与遗传因素密切相关。研究表明,牙齿的发育过程受到严格的基因控制,这些基因决定了牙齿的数量、形状和位置。多年来,通过转基因动物研究,我们获得了功能数据,证明骨形态发生蛋白(BMP)、成纤维生长因子(FGF)、音猬因子(SHH)和WNT信号通路的基因突变会导致牙齿发育异常,从牙齿形态缺陷到牙齿发育完全停止。
目前,已有多种基因突变被认为是TA的致病因素。然而,已知的突变仅能解释部分病例,因此关于TA的潜在分子机制仍有许多未知领域。最近,表观遗传调控也被认为在TA的发生中发挥重要作用。此外,新一代测序等先进技术的出现,为识别新型TA基因和变异提供了有力支持,也揭示了这些遗传缺陷的潜在病因。
2. 牙齿缺失的遗传基础
2.1 与牙齿发育不全相关的综合征
TA常常作为约150种综合征的一部分出现。这些综合征通常与口面部裂隙综合征和外胚层发育不良综合征相关。由于牙齿发育与其他外胚层器官的发育机制相似,参与牙齿发育的基因往往也在其他器官的发育过程中发挥作用。表1总结了与TA相关的综合征形式及其病因基因。
表 1.与综合征牙缺失及相关表型有关的基因。
|
基因/位点
|
OMIM | 染色体 | 综合症 | 遗传方式 | 牙齿/口腔表型 | 动物模型 | 动物模型表型 |
| ADAMTS2 | 604539 | 5q35.3 | 埃勒斯-丹洛斯综合征 | 常染色体隐性遗传 | 牙齿缺失、小牙、牙齿变色 | 牛模型 | 皮肤孢子病表型类似于 EDS VII C 型 |
| ANTXR1 | 606410 | 2p13.3 | 生长迟缓、脱发、假性无牙症和视神经萎缩 (GAPO) 综合征 | 常染色体隐性遗传 | 牙齿发育不全,萌出延迟 | 是的 | 生长迟缓、骨质流失、头骨缩短、额骨凸起、中面部发育不全 |
| AXIN2 | 604025 | 17q24.1 | 少牙-结直肠癌综合征 | 常染色体显性遗传 | 少牙症 | 是的 | 颅骨形态异常 |
| COL1A1/2 | 120150 | 17q21.33 | 成骨不全症 1 型 | 常染色体显性遗传 | 牙齿缺失、少牙 | 是的 | 致命、骨折 |
| CREBBP | 600140 | 16p13.3 | 鲁宾斯坦-泰比综合征 | 常染色体显性遗传 | 牙齿发育不全、下颌后缩、小颌畸形、腭弓/窄腭、爪尖、牙齿拥挤、螺丝刀门牙、交叉咬合和牙釉质发育不全 | 是的 | 骨骼畸形 |
| EDA | 300451 | Xq13.1 | 外胚层发育不良,少汗症 | 性连锁隐性遗传 | 无牙症、牙齿发育不全、牙齿畸形、小牙症 | 是犬 | 不完整的锥形牙齿 |
| EDAR | 604095 | 2季度13 | 外胚层发育不良,少汗/毛发/牙齿型 | 常染色体隐性遗传 | 无牙、缺牙、少牙 | 小鼠模型 | 臼齿数量减少、门牙小、臼齿小、釉质结形态异常 |
| EDARADD | 606603 | 1q42-q43 | 外胚层发育不良,少汗/毛发/牙齿型 | 常染色体显性遗传 | 无牙症、缺牙症、牛牙症、小牙症 | 小鼠模型 | 牙齿形态异常,臼齿数量减少,臼齿小,牙釉质形态异常 |
| EVC | 604831 | 4p16.2 | Ellis-van Creveld 综合征和 Weyers 肢端骨发育不全 | 常染色体隐性遗传/常染色体隐性遗传 | 原生牙、牙釉质异常、牙齿缺失、小牙畸形 | 小鼠模型 | 牙釉质缺陷、牙齿形态异常 |
| EVC2 | 607261 | 4p16.2 | Ellis-van Creveld 综合征和 Weyers 肢端骨发育不全 | 常染色体隐性遗传/常染色体隐性遗传 | 原生牙齿、牙釉质异常、牙齿缺失、少牙、小牙 | 小鼠模型 | 小牙畸形,上门牙较小,颅骨较小 |
| FGF10 | 602115 | 5p12 | 泪耳齿指综合征 | 常染色体显性遗传 | 牙齿发育不全(上颌门牙)、小牙畸形、延迟萌出、牙釉质发育不良 | 小鼠模型 | 牙齿形态异常,门牙短,臼齿小,腭发育异常,舌头形态异常 |
| FGFR1 | 136350 | 8p11.23 | 卡尔曼综合征 | 卡侬座 | 牙齿发育不全、唇裂/腭裂 | 小鼠模型 | 颅骨形态异常、面部不对称、门牙过长 |
| FGFR2 | 176943 | 10q26.13 | 泪耳齿指综合征 | 常染色体显性遗传 | 牙齿发育不全(上颌门牙)、小牙畸形、萌出延迟、牙釉质发育不良 | 小鼠模型 | 牙齿发育停滞、门牙过长、臼齿数量减少、小颌畸形 |
| Apert 综合征 | 常染色体显性遗传 | 牙齿发育不全(上颌犬齿)、牙釉质混浊、异位萌出、牙龈增生 | 小鼠模型 | 牙齿发育停滞、门牙过长、臼齿数量减少、小颌畸形 | |||
| FGFR3 | 134934 | 4p16.3 | 伴有黑棘皮症的 Crouzon 综合征 | 常染色体显性遗传 | 牙齿发育不全、错颌畸形、牙骨质瘤、牙齿萌出延迟、中面部发育不全 | 小鼠模型 | 牙齿错位、门牙过长、咬合不正、下颌突出、上颌后缩 |
| FLNB | 603381 | 3p14.3 | Larsen 综合征 | 常染色体显性遗传 | 牙齿发育迟缓、牙齿发育迟缓、3 类咬合、形态异常 | 小鼠模型 | 颅骨形态异常 |
| FOXC1 | 601090 | 6p25.3 | Axenfeld–Rieger 综合征 3 型 | 常染色体显性遗传 | 牙齿缺失、小牙畸形、牛牙畸形 | 小鼠模型 | 下颌骨短 |
| GJA1 | 121014 | 6q22.31 | 眼牙指发育不良 | AD、AR | 小牙畸形、牙釉质发育不全、牙齿发育不良、牙齿萌出延迟 | 小鼠模型 | 牙齿形态异常、小牙畸形、上下颌骨小、牙釉质厚度减少 |
| GRHL2 | 608576 | 8q22.3 | 外胚层发育不良/身材矮小综合征 | 常染色体隐性遗传 | 牙齿萌出延迟、牙齿发育不全、牙釉质发育不全 | 小鼠模型 | 颅骨形态异常、面部和中线裂隙 |
| IRF6 | 607199 | 1q32.2 | 范德沃德综合征 | 常染色体显性遗传 | 牙齿发育不全、唇裂/腭裂 | 小鼠模型 | 牙齿形态异常、腭部发育异常、下颌骨较小 |
| JAG1 | 601920 | 20p12.2 | 阿拉吉尔综合征 | 常染色体显性遗传 | 牙齿发育不全、牙釉质发育不全和混浊、矿化不足 | 小鼠模型 | 上颌骨短、咬合不正、腭形态异常 |
| KDM6A | 300128 | Xp11.3 | 歌舞伎综合症2 | XLR | 高腭弓、错颌畸形、小牙畸形、小牙弓、牙齿缺失、严重上颌骨萎缩、锥形牙齿 | 小鼠模型 | 颅裂 |
| KMT2D | 602113 | 12q13.12 | 歌舞伎症候群1 | 常染色体显性遗传 | 高腭弓、错颌畸形、小牙畸形、小牙弓、牙齿缺失、严重上颌后缩、锥形牙齿 | 小鼠模型 | 上颌骨短,吻部扁平 |
| KREMEN1 | 609898 | 22q12.1 | 外胚层发育不良,毛发/牙齿类型 | 常染色体隐性遗传 | 少牙、牙齿发育不全、牙槽嵴缺失、腭深度增加 | 是的 | 无颅面表型 |
| MKKS | 604896 | 20p12.2 | Bardet-Biedl 综合征 | 常染色体隐性遗传 | 牙齿拥挤、高腭弓、牙齿发育不全、错颌畸形、牙釉质发育不全、下颌后缩 | 小鼠模型 | 嗅觉上皮异常 |
| MSX1 | 142983 | 4p16.1 | Witkop 综合征 | 常染色体显性遗传 | 牙齿缺失、少牙 | 小鼠模型 | 牙齿发育停滞、指甲床和指甲板缺陷、腭裂 |
| NEMO | 300248 | Xq28 | 色素失调症 | 西洋参 | 缺牙、无牙、小牙 | 小鼠模型 | 无颅面表型 |
| NSD1 | 606681 | 5q35.3 | Sotos 综合征 I | 常染色体显性遗传 | 牙齿发育不全、牙釉质缺损、错颌畸形 | 小鼠模型 | 无颅面表型 |
| OFD1 | 300170 | 2.2 | 口面指综合征I | 性连锁遗传 | 牙齿发育不全、侧门牙缺失、犬齿错位、小颌畸形 | 是的 | 初级纤毛形成然后消失,肾囊肿 |
| P63 | 603273 | 3q28 | 颌面裂 8、拉普-霍奇金畸形、缺指症、外胚层发育不良和唇腭裂综合征 3 | 常染色体显性遗传 | 牙齿发育不全、牙釉质发育不全、广泛龋齿、下颌尖牙发育不全、普遍性小牙畸形、上颌恒切牙边缘脊突出、恒磨牙呈圆形、上颌恒中切牙呈桶状 | 小鼠模型 | 牙齿发育停滞、上下颌骨小、颅面发育异常、腭裂 |
| PITX2 | 601542 | 4q25 | Axenfeld–Rieger 综合征 1 型 | 常染色体显性遗传 | 牙齿发育不全、小牙畸形、牙釉质发育不全 | 小鼠模型 | 上颌骨和下颌骨形态异常,牙齿发育停滞 |
| PVRL1 | 600644 | 11q23.3 | 唇腭裂-外胚层发育不良 | 常染色体隐性遗传 | 牙齿缺失、唇腭裂、牙齿形态异常、小牙畸形 | 小鼠模型 | 牙齿形态异常 |
| RECQL4 | 603780 | 8q24.3 | 罗斯蒙德-汤姆森综合征 | 常染色体隐性遗传 | 牙齿发育不全、小牙、牙齿发育不全 | 小鼠模型 | 牙齿萌出延迟、腭裂 |
| RSK2 | 300075 | Xp22.12 | Coffin–Lowry 综合征 | 性连锁遗传 | 高窄腭、中线舌沟、牙齿缺失和小牙畸形 | 小鼠模型 | 牙齿形态异常、多生牙 |
| SHH | 600725 | 7q36.3 | 全前脑畸形 | 常染色体显性遗传 | 唇腭裂、单中央门牙、小颌畸形 | 是的 | 牙齿形态异常、小牙畸形 |
| TBX3 | 601621 | 12q24.21 | 尺乳综合征 | 常染色体显性遗传 | 牙齿发育不全、犬齿异位和发育不全 | 是的 | 继发性腭裂 |
| TCOF1 | 606847 | 5q32-q33 | 特雷彻·柯林斯综合征 | 常染色体显性遗传 | 牙齿缺失、小颌畸形、错颌畸形、牙齿间隙 | 是的 | 下颌骨和上颌骨较短 |
| TFAP2B | 601601 | 6p12.3 | 炭疽病 | 常染色体显性遗传 | 少牙、牙齿发育不全、厚嘴唇、乳牙滞留 | 是的 | 无颅面表型 |
| TFAP2B | 190685 | 21q22.13 | 唐氏综合症 | 零星病例 | 牙齿发育不全,萌出延迟,上颌中切牙呈桶状 | 是的 | 全身发育不全和发育迟缓、肾积水、心脏和神经系统缺陷 |
| TFAP2B | 612920 | 21q22.3 | 外胚层发育不良 | 常染色体隐性遗传 | 牙齿缺失、小牙 | 是的 | 无颅面表型 |
| UBR1 | 605981 | 15q15.2 | 约翰逊-暴风雪综合征 | 常染色体隐性遗传 | 少牙症 | 是的 | 无颅面表型 |
| WNT10A | 606268 | 2q35 | 牙髓发育不良 | 常染色体隐性遗传 | 少牙症、牙齿缺失症、小牙症 | 是的 | 磨牙发育停滞、多生磨牙、牙齿形态异常 |
| 肖普夫-舒尔茨-帕萨格综合征 | 常染色体隐性遗传 | 少牙症、牙齿缺失症、小牙症 | 是的 | 磨牙发育停滞、多生磨牙、牙齿形态异常 |
OMIM,人类在线孟德尔遗传(https://www.ncbi.nlm.nih.gov/omim/)AR,常染色体隐性遗传;AD,常染色体显性遗传;XLD,x 连锁显性遗传;XLR,x 连锁隐性遗传;IC,孤立病例。
2.1.1. 外胚层发育不良综合征
外胚层发育不良 (ED) 是一类表现为皮肤、毛发、指甲、牙齿、外分泌腺和皮脂腺等多个系统缺陷的疾病。已有研究揭示了由不同基因突变引起的各种ED类型,其中X连锁隐性少汗症 (HED) 是最常见的ED,主要由EDA基因突变引起。目前,已发现超过200种EDA基因突变。EDA蛋白是一种肿瘤坏死因子超家族成员,作为信号分子参与上皮形态发生。通过与其受体EDAR结合,EDA激活下游靶蛋白,调控相关细胞过程。EDAR与EDARADD基因突变会导致常染色体显性或隐性遗传的HED。除了牙齿发育受损外,ED还常伴随轻微的外胚层异常。
Witkop综合征是一种由佳学基因检测进行检测与分析常染色体显性ED,涉及牙齿和指甲异常,由MSX1基因突变引起。尽管少数病例中有毛发稀疏或细弱,大多数患者的毛发和汗腺正常。MSX1是一个转录因子,在脊椎动物发育过程中发挥关键作用,参与多个组织的发育。在小鼠中,MSX1对牙胚的初期发育至关重要,其缺失会导致牙齿发育停滞和颅面骨骼缺陷。超过20种MSX1突变与人类的综合征性和非综合征性TA相关。
牙甲皮发育不良 (OODD) 和Schopf–Schulz–Passarge综合征 (SSPS) 由WNT10A基因的纯合或复合杂合突变引起,是常染色体隐性遗传病。这些病症表现为干燥头发、严重的TA、光滑舌、掌跖角化病和指甲异常。WNT10A在牙齿发育中扮演重要角色,通过Wnt-β-catenin信号通路激活下游靶基因。尽管在小鼠中WNT10A敲除导致牙齿发育异常,但人类的TA表型却与之相反,表现为牙齿缺失。
在一些巴勒斯坦血缘家族中,KREMEN1基因的纯合突变被与ED相关联。KREMEN1作为Wnt信号通路的负调节因子,通过抑制Wnt信号的转导调节细胞过程。KREMEN1突变的小鼠表现为肢体缺陷和骨密度增高,但未见显著的其他表型。
2.1.2. 口面部裂隙综合征
范德沃德综合征 (VWS) 是最常见的唇腭裂综合征之一,具有常染色体显性遗传方式。患者通常有唇裂(可能伴腭裂),并可能出现TA。VWS由IRF6基因突变引起,IRF6在颅面发育过程中调节角质形成细胞的增殖和分化。小鼠模型显示IRF6缺失会导致皮肤、肢体和颅面发育异常,表现为腭裂等畸形。IRF6的突变是孤立性和综合征性唇腭裂的主要遗传因素。
唇腭裂-外胚层发育不良综合征是由PVRL1基因突变引起的一种由佳学基因检测进行检测与分析常染色体隐性遗传病。PVRL1编码的nectin-1在细胞间粘附和紧密连接的形成中发挥关键作用。临床上,患者可能表现为独特的面部特征、唇腭裂、外胚层发育不良、手指或脚趾并指畸形及牙齿发育异常。
2.1.3. Axenfeld–Rieger综合征
Axenfeld–Rieger综合征是一种常染色体显性遗传病,表现为眼前节发育异常,约50%的患者因青光眼失明。此病还可能伴随全身异常,如脐带复旧不全、尿道下裂及牙齿异常。该病由PITX2基因突变引起,该基因在牙齿发育过程中发挥重要作用。小鼠研究表明,PITX2缺失导致牙齿发育停止,并在脊椎动物的左右不对称形成中扮演关键角色。
2.1.4. 家族性腺瘤性息肉病综合征
家族性腺瘤性息肉病 (FAP) 是一种常染色体显性遗传病,特征为结肠和直肠的多发性腺瘤性息肉。该病还可能伴随表皮样囊肿、纤维瘤和牙齿异常,包括多生牙和复合牙瘤等。FAP由APC基因突变引起,约17%的患者有牙齿异常,部分病例出现TA。研究表明,FAP患者的牙齿异常可能与结肠癌的遗传易感性相关。
2.1.5. 少牙-结直肠癌综合征
AXIN2基因的种系突变与芬兰一个家族的少牙症和结直肠癌密切相关。该家族的成员有多达8颗恒牙缺失。研究还发现AXIN2突变与结直肠癌相关基因变异密切相关,提示这些基因可能在牙齿发育及肿瘤发展中发挥作用。
2.2. 孤立性(非综合征性)牙齿缺失
动物模型中综合征性缺牙(TA)的综合征形式和与正常牙齿发育有关的基因为识别人类孤立性综合征性缺牙(TA)的候选基因提供了重要线索。迄今为止,已有许多基因被提出为孤立性综合征性缺牙(TA)的病因(表 2)。
表 2.:与孤立性牙齿发育不全有关的基因。
2.2.1. MSX1
MSX1突变是孤立性综合征性缺牙(TA)患者中首次描述的突变 [ 152 ]。此后,已报道了 20 多种与孤立性综合征性缺牙(TA)相关的MSX1突变,其中大多数是位于同源框结构域的无义突变或错义突变,这表明MSX1的单倍体不足是综合征性缺牙(TA)表型的基础 [ 153 ](图 1)。同源框结构域的突变会破坏 DNA 结合并优先导致孤立性 TA,同时蛋白质天然未折叠的 N 端部分的变异通常会导致口面部裂。这些观察结果表明,MSX1 突变的影响与受影响的蛋白质结构域直接相关。MSX1相关综合征性缺牙(TA)通常包括缺失上颌和下颌第二前磨牙以及上颌第一前磨牙。
图 1.
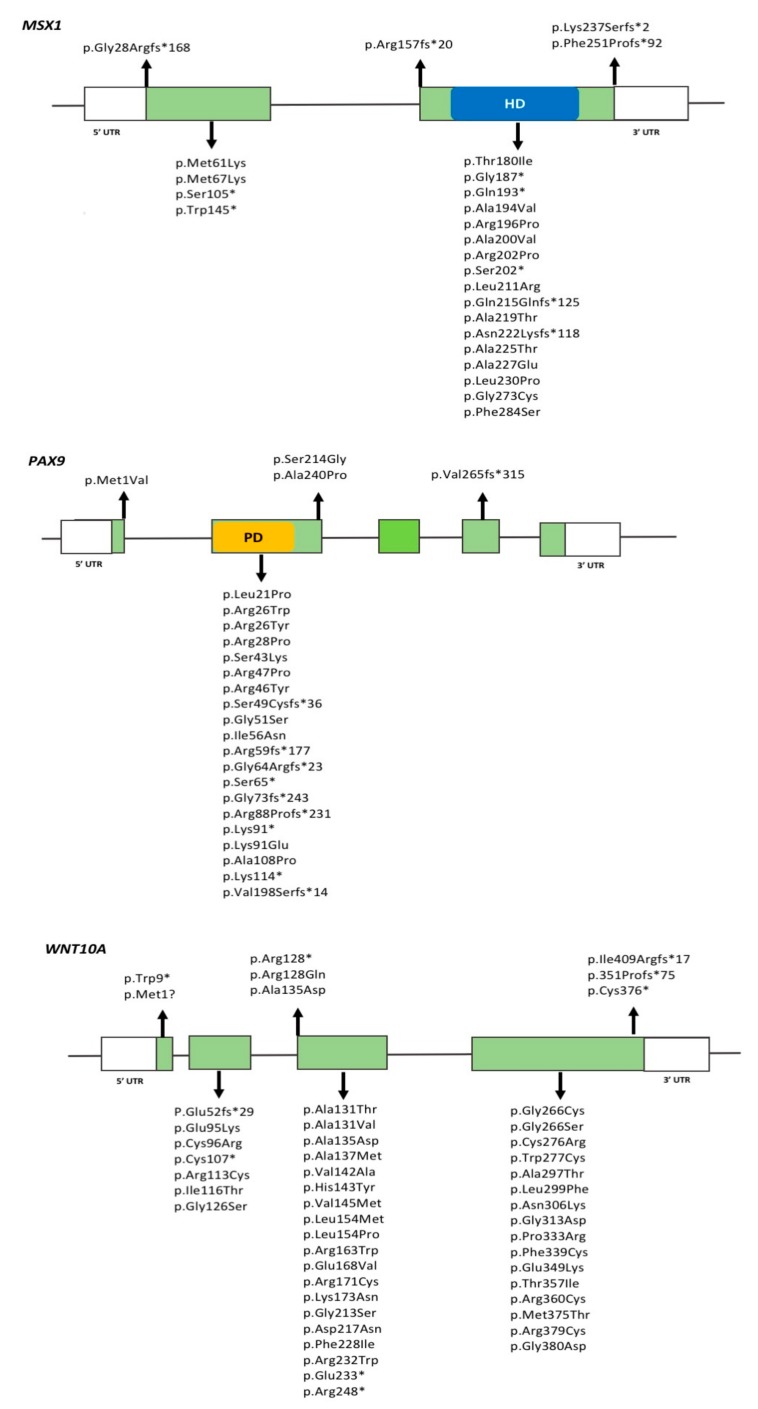
MSX1、PAX9和WNT10A基因中预测的错义、移码和无义突变的位置。绿色框代表外显子,外显子之间的水平线代表内含子。HD 对应于MSX1中的同源域。PD 对应于PAX9中的配对域。UTR:非翻译区。
2.2.2. PAX9
PAX9 属于成对盒 (PAX) 转录因子家族,这个家族对多种多细胞生物的正常发育至关重要。除了 MSX1 外,PAX9 长期被认为与分离型综合征性缺牙(TA)表型相关,且是牙齿发育研究中最广泛研究的基因之一 。PAX9 在假定的牙间充质中表达,起到激活信号并启动牙齿发育的作用。在小鼠模型中,PAX9 的缺失会导致牙齿发育在芽期停止。
迄今为止,已收录了30多种与综合征性缺牙(TA)相关的 PAX9 变异,其中大多数位于基因外显子2,表现为插入/缺失或错义突变,这些变异影响了 PAX9 蛋白的配对结构域 (见图1)。PAX9 变异通常与第二恒磨牙的发育不全有关,其次是第二前磨牙,少数情况下也与前牙发育不全相关。总体而言,TA 表型的严重程度与突变类型及其对 PAX9 功能的影响密切相关。与错义突变的个体相比,携带无义突变或移码突变的个体通常表现出更严重的表型。在综合征性缺牙(TA)中,已知的 PAX9 突变呈杂合遗传,符合常染色体显性遗传模式,表明单倍体不足可能是导致该表型的原因。有研究表明,携带 PAX9 突变的综合征性缺牙(TA)患者通常表现出较小的牙冠尺寸。
2.2.3. AXIN2
AXIN2 的稀有和常见变异与孤立型综合征性缺牙(TA)有关,且常表现为受影响牙齿的混合发育模式。在综合征性缺牙(TA)个体中,已描述了磨牙、下切牙和上侧切牙的发育不全,并且至少有一颗切牙缺失的情况较为常见。文献中广泛报道了五种 AXIN2 突变,包括四种错义突变(c.956+16A > G;p.Pro50Ser、c.2051C > T;p.Ala684Val、c.2062C > T;p.Leu688Leu 和 c.2272G > A;p.Ala758Thr)和一种移码突变(c.1994insG;p.Asn666GlyfsX41)。在所有受影响的个体中,移码突变与牙齿缺失的关联性高于错义突变。
2.2.4. WNT10A
WNT10A 一直是许多综合征性缺牙(TA)遗传研究的重点。在缺失1至3颗牙齿的综合征性缺牙(TA)患者中,约 15.8% 的病例以及缺失超过4颗牙齿的综合征性缺牙(TA)患者中,约 52% 的病例已发现 WNT10A 的 50 多种杂合、纯合及复合杂合变异 。最近的基因型-表型相关性研究为 WNT10A 在综合征性缺牙(TA)中的作用提供了深入的见解。总体而言,与仅携带单一变异的个体相比,WNT10A 的复合杂合突变与严重的综合征性缺牙(TA)和大量牙齿缺失相关。尽管 WNT10A 变异个体没有特定的牙齿缺失模式,但上颌和下颌磨牙以及下颌前牙缺失的情况经常被报。值得注意的是,杂合的 WNT10A 变异也已在未患综合征性缺牙(TA)的家庭成员以及无综合征性缺牙(TA)或无综合征性缺牙(TA)家族史的对照个体中发现。据估计,携带 WNT10A 单一杂合变异的个体中,约 41% 不会患上 TA。
图1 展示了在综合征性缺牙(TA)患者中发现的 WNT10A 变异。一些 WNT10A 变异被认为是特定人群中常见的“热点”突变。例如,c.637G > A (p.Gly213Ser) 变异在亚洲人群中较为常见,而 c.682T > A (p.Phe228Ile) 变异则在白种人群体中的综合征性缺牙(TA)患者中以纯合或杂合形式广泛报道,且在正常对照中也有 2.3% 的频率。Phe228Ile 变异是最常见的变异,通常与 WNT10A 或其他基因中的变异共同出现。这些发现支持综合征性缺牙(TA)的寡基因遗传模型,后文将对此进行讨论。
2.2.5. LRP6
LRP6(LDL 受体相关蛋白 6)是 Wnt/β-catenin 信号通路中的辅助受体,最近有研究表明它在孤立型综合征性缺牙(TA)的发病中起到一定作用。在患有散发性综合征性缺牙(TA)或家族中有综合征性缺牙(TA)分离现象的个体中,发现了六种 LRP6 变异,包括一个无义变异(c.1779dupT,p.Glu594*)、两个插入突变(c.2224_2225dupTT,p.Leu742Phefs7 和 c.1144_1145dupAG,p.Ala383Glyfs8)以及一个剪接位点突变(c.3607+3_6del,p.?),这些突变均导致截短的 mRNA 产物,此外还有一个错义突变(c.56C > T,p.Ala19Val)。在小鼠中,LRP6 在牙囊和内釉质上皮中表达,而 LRP6 的纯合缺失会导致严重的骨骼异常并且致死。
2.2.6. 近期与综合征性缺牙(TA)相关的其他基因
在 7 名患有 TA(牙齿发育不全)和其他畸形(如牛牙畸形、毛发稀疏且生长缓慢、皮肤干燥和瘙痒)的患者中发现了 GREM2(编码 GREMLIN2)的突变 。GREMLIN2 被知晓在胚胎发育过程中调节 BMP,其中 BMP4 在牙齿发育中起着重要作用,且其敲除会导致小鼠牙齿发育停滞。有趣的是,尽管 GREM2 基因敲除小鼠的牙齿小且畸形,但牙齿发育并未停滞。这些发现表明 GREM2 在牙齿发育过程中可能发挥着作用。GREM2 中的三种错义突变(p.Ala13Val、p.Glu136Asp 和 p.Gln76Glu)已被鉴定为与单独患综合征性缺牙(TA)的个体相关的致病变异,且尚未与其他结构畸形相关。即便在同一家族中,GREM2 突变的表现度也有所不同。
EDA、EDAR 和 EDARADD 也被认为与孤立型综合征性缺牙(TA)相关。在一项基因型-表型相关性研究中,孤立型综合征性缺牙(TA)患者中的所有 EDA 突变均为错义突变,并且最可能出现在 TNF 结构域。
另一个与 WNT 通路相关的基因 WNT10B(无翅型 MMTV 整合位点家族,成员 10B)也与孤立型综合征性缺牙(TA)相关,尽管这种变异主要出现在中国和泰国的家族中。在中国的综合征性缺牙(TA)患者中,尤其是缺失上侧门牙的个体中,发现了 WNT10B 中的三个杂合错义突变(c.632G > A,p.Arg211Gln;c.569C > G,p.Pro190Arg;和 c.851T > G,p.Phe284Cys)以及一个无义突变(c.786G > A,p.Trp262*)。最近在泰国的五个家族中,发现了两个杂合错义突变(c.475G > C,p.Ala159Pro 和 c.1052G > A,p.Arg351His),并与单独的综合征性缺牙(TA)以及小牙畸形和牛牙畸形等其他牙齿异常相关。
ANTXR1(炭疽毒素受体1)的纯合错义变异 c.1312C > T(p.Arg438Cys)已被证实与土耳其一家庭的 TA(少牙症)相关。ANTXR1 的纯合和双等位基因突变与生长迟缓、脱发、假无牙症和视神经萎缩(GAPO)综合征相关,这些症状属于常染色体隐性遗传特征。在小鼠中,有针对性地破坏 Antxr1 可获得无重大结构缺陷的存活小鼠,但观察到牙齿过度生长、门牙错位和牙齿发育不良。在牙齿发育的早期阶段,Antxr1 的表达存在于发育中的舌、上颌和下颌突的上皮、牙齿上皮及间充质中,后期则出现在釉质器上皮和牙乳头中,并最终转移至成釉细胞的极化层和正在分化的成牙本质细胞中。ANTXR1 是一种与结直肠癌相关的肿瘤特异性内皮细胞标志物,在肿瘤血管生成过程中上调。此前,Lammi 等人 发现肿瘤抑制基因 AXIN2 的突变与一例患有严重 TA(少牙症)和结直肠癌的家族相关,并提出综合征性缺牙(TA)和结直肠癌可能具有共同的遗传病因。此后,已有大量研究报道与综合征性缺牙(TA)相关的癌症基因变异。这些发现凸显了综合征性缺牙(TA)的复杂性,并强调在研究中考虑修饰基因和/或基因间相互作用的必要性。
最近的一项全基因组关联研究(GWAS)纳入了 1900 多例综合征性缺牙(TA)病例和 33 万例具有欧洲血统的对照,发现了 4 种与综合征性缺牙(TA)相关的新风险变异,以及 5 种与综合征性缺牙(TA)伴随口面部腭裂的综合表型相关的风险变异 。口腔异常是口面部腭裂儿童的常见症状,人们根据相关口腔异常的模式提出了腭裂亚表型。在这9个变异中,5个位于或接近与牙齿发育和/或其他外胚层结构发育有关的 Wnt 通路基因(EDA、EDAR、FOXI3、FORXP1 和 LEF1),其余 4 个则位于或接近与综合征性缺牙(TA)或牙齿发育无关的基因(ASCL5/CACNA1S、ARHGAP15、NOL11 和 FAM49A)。此外,本研究还发现 WNT10A 中的两个已知变异(p.Phe228Ile 和 p.Cys107*)与综合征性缺牙(TA)显著相关。
3. 单基因遗传模型与寡基因遗传模型
近年来,针对多种孟德尔疾病,研究者提出了寡基因遗传和多位点变异模型,并进一步确立了突变负荷的概念。对于TA(少牙症),寡基因遗传的证据逐渐显现,这得到了最近全外显子组测序研究和对多个候选基因进行直接测序研究结果的支持。
研究表明,MSX1和PAX9的双基因突变与更严重的TA表型(15至17颗牙齿缺失)相关。这些基因之间的相互作用已经初步揭示:PAX9与MSX1共同作用,协同激活下游牙齿发育基因(如BMP4)的表达,这对于牙齿形态发生至关重要。当这两个基因发生双基因突变时,它们的相互作用可能会被破坏,从而导致更严重的TA表型。
最近的研究还发现,TA患者中有些人表现出WNT10A的双等位基因或杂合基因型,并同时携带EDA、EDAR或EDARADD基因的纯合或杂合突变,这表明不同基因中等位基因的综合效应导致了TA。此外,在一名上颌恒尖牙TA患者中,发现了WNT10A的复合杂合突变(IVS2 + 1G > A和c.637G > A)以及GREM2的错义杂合变体(c.38C > T)。
在一个土耳其近亲家族中,还发现了DKK1(Dickkopf WNT信号通路抑制因子1;c.548-4G > T)和COL17A1(XVII型胶原蛋白α1链;c.3277 + 3G > C)中的杂合剪接突变,以及LAMA3(层粘连蛋白α3亚基;c.2798G > T)中的杂合错义变体与TA相关。这些基因的致病突变尚未在TA患者中广泛发现,但由于它们的生物学作用和/或与疾病表型的相关性,它们可以被视为潜在的候选基因。DKK1编码高亲和力的Dickkopf同源物1跨膜受体,能够与LRP6协同作用,在发育及其他细胞过程中阻断Wnt信号传导。在小鼠中,DKK1在牙齿间充质、成牙本质细胞和成骨细胞中表达,其转基因小鼠胚胎口腔上皮中的异位表达会阻断上皮和间充质的信号传导,导致牙齿发育在芽期停止。此外,DKK1的常见单核苷酸多态性(rs11001553)已被发现在中国汉族人群中与单独的TA相关。
LAMA3基因突变与交界性大疱性表皮松解症(OMIM #226650)相关,这是一种常染色体隐性皮肤病,特征包括多发水疱、糜烂、营养不良性指甲、牙釉质发育不全和牙齿发育不全。在小鼠中,LAMA3的靶向破坏会导致成釉质细胞分化缺陷。在患有牙釉质缺陷的大疱性表皮松解症患者中,也发现了COL17A1的突变。
此外,在一对意大利兄妹中,发现了PITX2基因5'UTR中的纯合变异(c.-387delC > G),该变异与Axenfeld–Rieger综合征相关,以及BMP4中的纯合错义变异(c.T455C,p.Val152Ala),这些变异与单独的TA一起分离。
多个基因座中的潜在致病等位基因的发现表明,孤立型TA可能存在寡基因遗传和多位点变异模型,这可能导致表型的多样性。随着对特征明确的TA个体和家族进行全基因组测序,并仔细研究基因型-表型相关性,可能会发现新的TA相关基因,这些基因可能单独作用或与其他基因相互作用。
4. 遗传途径是未来研究的重点
多年来,使用转基因动物的研究表明,BMP、FGF 和 WNT 信号通路基因缺陷会导致严重的牙齿发育异常,范围从牙齿形态缺陷到牙齿发育完全停止 。同时,尚未发现与综合征性缺牙(TA)相关的 FGF 家族基因突变,而在一个综合征性缺牙(TA)家族中发现了BMP4的单一变体(见上文)。
目前的现有证据支持 WNT 通路基因在孤立性综合征性缺牙(TA)中发挥重要作用,最主要的是TA 个体中AXIN2、WNT10A、WNT10B和LRP6的致病突变频率较高。WNT 信号分子对于胚胎发育过程中多种细胞类型的模式化、增殖和分化至关重要。据报道,牙上皮分泌 WNT,特别是WNT4、WNT6和WNT10,对牙齿发育至关重要,因为 WNT 信号的缺失会导致釉质结功能障碍,随后导致牙齿发育停滞。
EDA、EDAR和EDARADD在综合征型和孤立型综合征性缺牙(TA)中均发挥作用,它们属于 NF-kB 信号通路。NF-kB 通路中的其他基因包括NEMO(核因子 κB 激酶亚基 γ 抑制剂,一种重要的通路调节剂)和TRAF6(TNF 受体相关因子 6),尽管人们对这些基因在牙齿发育中的确切作用知之甚少,并且据报道,在综合征型综合征性缺牙(TA)患者中这两个基因存在变异 。
基于上述观察,多位点变异是某些综合征性缺牙(TA)病例的潜在解释,候选基因属于相同或不同的信号通路,可以假设单独的综合征性缺牙(TA)可能是多个基因变异的结果,这些基因单独作用或与其他基因结合作用,导致该疾病的不同表现度。确定综合征性缺牙(TA)中假定缺陷基因的全部范围、它们所属的通路(图 2)、它们的功能和相互作用伙伴,将有助于我们更好地了解综合征性缺牙(TA)的潜在机制,并可能成为未来预防和牙齿替换策略的基础。
图 2.
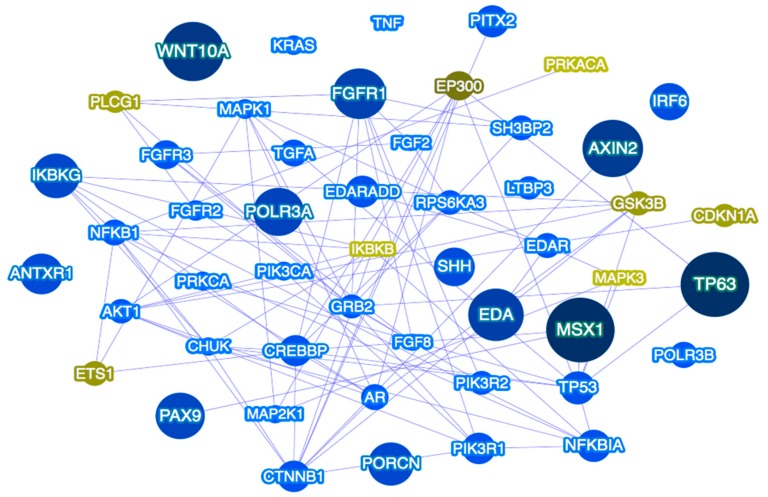
Phenolyzer 预测的牙齿发育不全基因网络。所示网络包括前 50 个优先基因及其与种子基因的预测关系。较大的深蓝色节点表示种子基因,中等宝蓝色节点表示相互作用基因。绿色字体表示预测基因。蓝线表示蛋白质-蛋白质相互作用。
5. 结论
孤立性综合征性缺牙(TA)是一种异质性疾病,其表现形式各异。虽然许多基因的变异被认为是综合征性缺牙(TA)的病因,但许多个体的综合征性缺牙(TA)病因仍未解决,可能反映了牙齿发育中未知的基因突变或多位点变异的存在。此外,环境和表观遗传因素也可能被视为综合征性缺牙(TA)表型的可能因素,应在未来的研究中加以探索。对特征明确的个体和家族进行下一代测序研究具有独特的能力,可以识别整个基因组中所有综合征性缺牙(TA)易感变异,同时揭示可能对牙齿发育至关重要的重要遗传和网络相互作用。进一步的遗传和功能研究侧重于新发现的基因和途径,有可能阐明孤立性综合征性缺牙(TA)的遗传图景,并为预防和治疗策略提供见解。针对综合征性缺牙(TA)相关基因和/或途径的靶向疗法可能代表未来的牙齿替代疗法。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
