












【佳学基因检测】CHILD综合征基因解码、基因检测有什么用?
遗传病、罕见病基因检测导读:
CHILD综合征是英文congenital hemidysplasia with ichthyosiform erythroderma and limb defects的中文翻译。这一疾病又叫做CHILD syndrome Ichthyosiform erythroderma, unilateral, with ipsilateral malformations, especially absence deformity of limbs,Child Syndrome、 Ichthyosiform Erythroderma, Unilateral, with Ipsilateral Malformations, Especially Absence Deformity of Limbs、 Congenital Hemidysplasia with Ichthyosiform Nevus and Limbs Defects、 Child Nevus、 Congenital Hemidysplasia with Ichthyosiform Nevus and Limb Defects、 Ichthyosis, Child Syndrome、 Child Syndrome Ichthyosis、 Child。该病是一种生殖障碍方面的基因病、遗传病。佳学基因通过基因解码找到了导致这一疾病发生的基因。可以通过基因检测阻止CHILD综合征在后代或者二胎中的出现。根据《人的基因序列变化与人体疾病表征》,该病属于代谢病、发育异常和生殖障碍基因病。
什么样的人应当做CHILD综合征基因解码、基因检测?
先天性半发育不良伴鱼鳞状红斑和肢体缺陷,通常用缩写CHILD综合征来称呼,是一种影响身体多个部位发育的疾病。该疾病的症状通常仅限于身体的右侧或左侧。(“Hemi-”表示“半”,“dysplasia”表示异常生长。)右侧比左侧受影响的可能性高两倍。 患有CHILD综合征的人会有一种皮肤病,表现为皮肤大面积红肿和覆盖有脱屑的鳞片状(鱼鳞痣)症状。该病通常出现在皮肤皱褶和皱纹处,但通常不影响面部。这种皮肤异常在出生时就已经存在,并持续终身。 CHILD综合征还会在早期发育阶段干扰手臂和腿的形成。患有这种疾病的儿童可能出生时一条或多条肢体短缩或缺失。肢体异常出现在身体同侧的皮肤异常上。 此外,CHILD综合征还可能影响大脑、心脏、肺和肾的发育。先天性半发育不良伴鱼鳞状红斑和肢体缺陷(CHILD)综合征(OMIM编号:308050)是一种伴有病理性突变的NAD(P)依赖性类固醇脱氢酶样蛋白(NSDHL)基因(OMIM编号300275)的鱼鳞病。这个将近39 kb的基因位于X染色体的q臂末端附近,编码参与胆固醇生物合成的酶。由于在男性胎儿中携带生殖系突变会导致死亡,因此该病几乎仅在女性中发现。 大多数CHILD综合征患者出生时表现出涉及身体一侧躯干和四肢的广泛鱼鳞状红斑,可以是弥漫性的或遵循Blaschko线路或两者兼有,并有明显的中线分界。黄色蜡状鳞片、翻盖性(对身体皱褶具有亲和力)和疣状黄色瘤的发生是额外的鉴别特征。常常也观察到同侧的骨骼畸形,范围从某些手指的发育不良到有效缺失的肢体。皮肤鱼鳞状红斑下方的其他器官也可能受到影响。该病的轻度和不太广泛的表现可能会在儿童早期时导致误诊。

佳学基因congenital hemidysplasia with ichthyosiform erythroderma and limb defects基因解码、基因检测病例分享
病例介绍:
这位佳学基因合作医疗机构送检的患者于23岁时到皮肤科诊,因为左侧后耳区域出现瘙痒和糜烂的皮疹已有2年时间。检查发现,有多个色素沉着、结痂和过度角化的丘疹(图1a)。这些皮损的皮肤镜检显示棕色网格状图案和黄色鳞屑(图1b)。此外,在她左前大腿和小指背部呈线性分布,存在黄褐色、鳞状、过度角化的斑块(图1c,d),以及左臂内侧和腋窝区域呈线性分布的色素减退条纹(图1e)和下背部的色素消退斑。在她婴儿期时,曾经在其他三级医疗机构被诊断为色素失调症,她的父母提供的发病显示,她出生时上述区域受到水疱的影响。这些水疱随后消退,导致了过度角化的斑块和色素减退斑块。她母亲的皮肤在两臂内侧和右前臂上也有类似的线性色素减退斑块。

图1:该患者不同区域皮损的影像:(a)左侧后耳区域出现多个色素沉着、结痂和过度角化的丘疹。(b)左侧后耳区域丘疹的皮肤镜图像。(c)左前大腿上存在黄褐色、鳞状、过度角化的线性斑块。(d)左小指背部存在黄褐色、鳞状、过度角化的线性斑块。(e)左臂内侧出现线性色素减退斑块。
该患者的左侧后耳区域和大腿的斑块组织活检显示表皮过度角化、柱状过角化、表皮葡萄状细胞增生和乳头层真皮内有多个黄色瘤细胞(图2a-d)。同时,她还向胃肠科医生反映反复出现的消化不良和心灼感。食管胃十二指肠镜检查显示近端十二指肠、胃和远端食管有多个白色斑块(图3a-e)。这些区域的活检显示类似的细胞呈现精细的空泡形态,位于基底物质内,证实其是巨噬细胞系的,并诊断为胃肠黄色瘤病(图4a-c)。这些细胞对CD163染色呈阳性(图4d),证实它们的巨噬细胞来源。
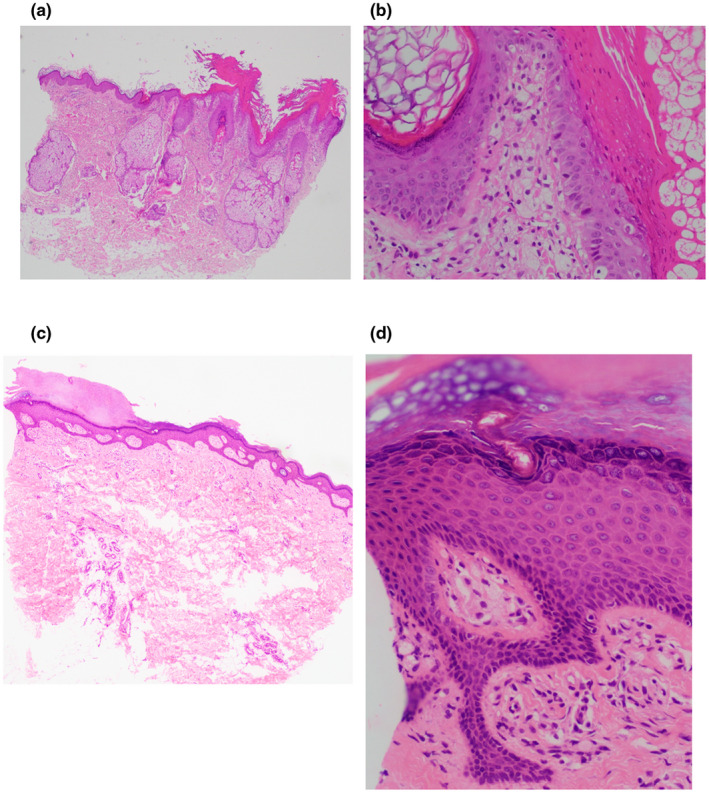

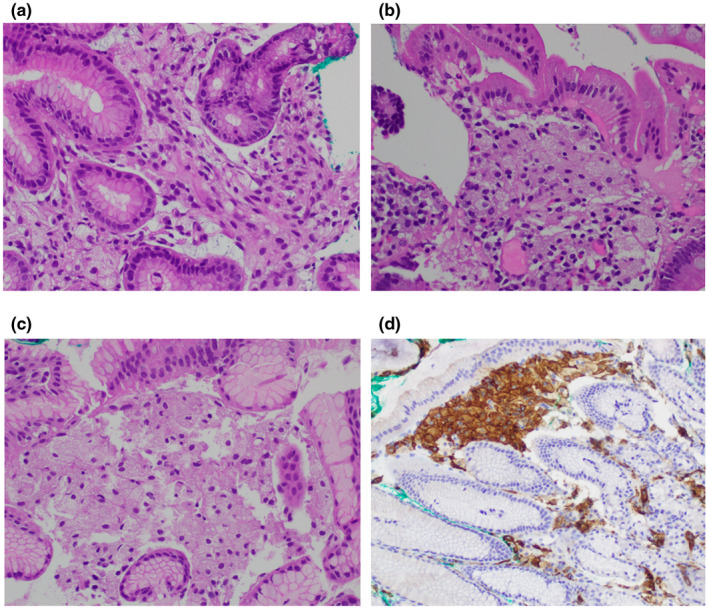
图4:400倍镜下胃肠道切片的苏木精-伊红染色:(a)胃窦区域显示层状结构内多个黄色瘤细胞。 (b)十二指肠区域显示层状结构内多个黄色瘤细胞。 (c)Z线区域黏膜显示层状结构内多个黄色瘤细胞。 (d)CD163(200倍)染色的Z线区域黏膜切片显示层状结构内多个巨噬细胞系黄色瘤细胞。
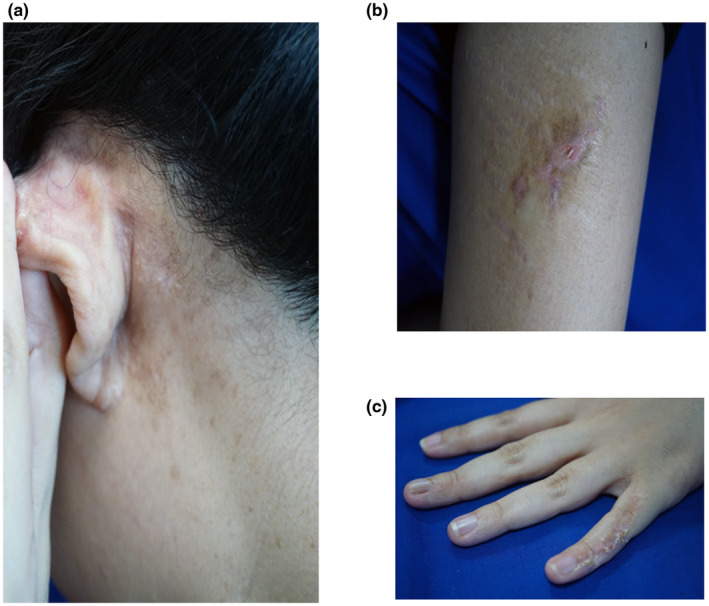
图5:患者使用复合配方乳膏(含1%辛伐他汀和2%胆固醇)治疗3个月后在皮肤上的效果,包括(a)左侧耳后区域,(b)左前股部和(c)左小指。
在进行临床病理相关性分析后,怀疑为CHILD综合症,并通过分子检测予以确认。患者开始使用含有1%辛伐他汀和2%胆固醇的复合配方乳膏治疗,并在治疗3个月后取得了良好的疗效(图5a-c)。她的胃肠道症状在使用质子泵抑制剂和抗酸剂治疗后有所改善。
致病基因鉴定基因解码的病例分析
从患者左侧耳后区和左大腿的皮损处采集穿刺活检标本。从患者和她的父母采集静脉血。使用DNeasy Blood and Tissue Kit (Qiagen Inc.,美国)和PureGene Blood Kit (Qiagen Inc.,美国)从皮肤组织和静脉血中提取DNA。使用Illumina's TruSight One试剂盒进行靶向面板测序,以从大腿组织提取的DNA为基础,发现NSDHL基因第3外显子中G核苷酸的重复插入,从而导致在下一个密码子中发生氨基酸变化的错义突变 (NM_015922.3: c.131dupG: p. Gly45Trpfs*24)。通过基于目标的Sanger测序验证了这个变异,并从皮肤组织和血液中分别测试了DNA。父母的DNA测试表明是母系遗传,患者的父亲检测结果为阴性。
重复插入的G核苷酸会导致错义突变,从插入点的下游24个密码子处发生早期终止。该变异在任何患者或人群数据库中均未被报道。然而,在一个菲律宾女孩身上曾报道过同一个密码子的错义突变(c.130G>A:p. Gly44Ser),被研究者认为是“可能致病”的。佳学基因病案集中收录了这一个罕见的CHILD综合征病例,该病例仅在成年后经过临床、病理和分子相关和确认后才被诊断。她的皮肤病变从出生时就存在,但只在成年后变得更加明显。这些早期的临床特征并没有考虑到CHILD综合征的诊断。相反,她被诊断为具有色素不正常的病变。目前,她皮肤病变的主要鉴别诊断是鳞屑性毛囊角化症。但是,ATP2A2和IKBKG的变异都没有被确定,排除了鳞屑性毛囊角化症和具有色素不正常的病变。该病例说明,CHILD综合征的临床谱可以包括具有细微的皮肤特征和几乎没有肢体缺陷的患者,需要皮肤活检以确认与多发性黄色瘤细胞相关的皮肤病变存在其他典型特征,如角化过度、正角化或表皮增生。
该病例的患者还出现了胃肠道症状,这在CHILD综合征病例中很少见,但也可能提供诊断线索,因为胃和十二指肠活检中存在类似的黄色瘤细胞。胃肠道广泛分布的黄色瘤细胞被认为是由于黄酮酶样蛋白依赖性类固醇脱氢酶样蛋白基因突变导致的毒性脂质代谢产物的沉积引起的粘膜炎症、损伤或创伤的反应性(Mavilia和Wu,2018)。胃肠道中的黄色瘤也与癌症、淋巴瘤和巨细胞病毒结肠炎有关。这一病例的数据库分析的意义在于将CHILD综合征添加到了胃肠道中多发性黄色瘤的鉴别诊断中。
congenital hemidysplasia with ichthyosiform erythroderma and limb defects致病鉴定基因解码
NSDHL基因的突变可以导致CHILD综合征。该基因的正常基因序列在体内指导合成与生殖、发育有关的关键酶,该酶参与胆固醇的生产。胆固醇是一种在身体内产生并从动物食物中获得的脂肪,特别是蛋黄、肉类、鱼类和乳制品。尽管高胆固醇水平是心脏病的众所周知的风险因素,但身体需要一定量的胆固醇来在出生前后正常发育和正常运作。胆固醇是细胞膜和神经细胞保护物质(髓鞘)的重要组成部分。此外,胆固醇在某些激素和消化酸的生产中也起着作用。 导致CHILD综合征的突变消除了NSDHL酶的活性,破坏了细胞内正常胆固醇的生产。缺乏这种酶可能还会导致胆固醇生产的潜在有毒副产物在身体组织中积累。NSDHL基因编码3β-羟基甾体醇脱氢酶,是胆固醇生物合成过程中的一个酶。由于胆固醇是表皮角质层中层片状结构的主要成分之一,其不足会导致这些层片状结构的破坏。类固醇中间产物的积累也可能导致异常的Hedgehog信号通路,导致类似CHILD综合征患者骨骼畸形的出现。含有胆固醇和他汀类药物(例如2%洛伐他汀/2%胆固醇)的复合外用剂治疗CHILD综合征皮肤病变的成功表明,皮肤病变不仅是由于胆固醇不足,而且是由于有毒代谢物的积累。基于基因解码结果的正确治疗的患者的改善进一步证明了这种复合外用剂治疗CHILD综合征皮肤病变的有效性。
自从NSDHL基因的突变被确定为CHILD综合症的潜在原因以来,没有其他基因与该疾病有关联。另一种与NSDHL相关的疾病是CK综合症(OMIM#300831),其特征是认知能力障碍、癫痫、小头畸形、大脑皮层畸形和畸形面部特征。CK综合症是一种影响大多数男性的X连锁隐性遗传病。相比之下,由于X连锁显性,CHILD综合症几乎有效影响女性。只报道了两例受影响的男性,他们很可能是由于体细胞后期突变而不是生殖细胞突变。另一种相关疾病,X连锁性骨软骨发育不全2型(CDPX2,OMIM#302960),可以表现出与CHILD综合症相似的临床特征,其归因于EBP(emopamil结合蛋白)基因的突变,该基因编码NSDHL通路下游的蛋白质。除了分布呈线性或螺旋状的鱼鳞病损外,这些患者还表现出近端骨骼和大腿骨的骨化点和软骨的放射学特征。
佳学基因的基因突变与疾病表征数据库在CHILD综合征患者中发现了30多个不同的致病性变异(包括插入和删除),涵盖了NSDHL基因。其中大多数是单核苷酸替换,其中包括13个错义突变、七个无义突变、两个剪接位点和一个3'非翻译区的替换,已记录在人类基因突变数据库中。也已经描述了多外显子缺失和小插入。在报告的大多数患者中,有的是欧洲血统,有的是为东亚人,包括中国人和日本人,还有其他地方的孤立案例。本例的另一个不寻常特征是母源性遗传,而大多数情况都是散发性的新生变异。
尽管CHILD综合征的皮肤表现已知限于身体的一侧,但也已有报道双侧表现的病例。大约有三分之二的患者表现为右侧受累,而左侧受累则与更严重的表型或早期死亡相关。该病例中的患者左侧受累,但其表型非常轻微,以至于一度怀疑为莫西卡病变。与两名其他携带移码变异的患者相比,该患者表现出更大的临床表型变异,包括骨骼异常和持续性红皮病灶。另一名携带与我们的患者相同密码子的错义变异的患者也表现出了典型的CHILD综合征症状。综合征表现的变异性表明表型展现受到表观遗传和环境因素的影响。
由于NSDHL基因位于X染色体上,表型严重程度变异的另一个合理解释是X染色体失活程度的偏差,可以在两个X染色体中的每一个之间在34%到68%之间变化。然而,这种解释可能性较小,因为NSDHL蛋白似乎对于细胞的生存至关重要:在体外培养的Nsdhl-Null成纤维细胞在48小时后即不能生存,还有证据表明在小鼠各种组织中存在负选择反对NSDHL缺陷细胞。
congenital hemidysplasia with ichthyosiform erythroderma and limb defects基因解码如何帮助婚恋和二胎生育
该病症具有X连锁显性遗传方式。如果导致该疾病的突变基因位于X染色体上,即两个性染色体中的一个X染色体,则该疾病被认为是X连锁的。如果每个细胞中的一个改变的基因副本足以引起该病,则该遗传方式为显性遗传。 大多数CHILD综合征病例是散发性的,这意味着家族中只有一个成员受到影响。很少情况下,该病会在家族中遗传并由母亲传给女儿。研究人员认为,CHILD综合征几乎只在女性中发生,因为受影响的男性会在出生前死亡。只有一个男性患有CHILD综合征的报告。这种疾病以常染色体隐性模式遗传,这意味着受累者每个细胞中的基因的两个拷贝都具有突变。常染色体隐性病患者的父母各携带一个拷贝的突变基因,但他们通常不显示病症的体征和症状。
CHILD综合征的数据库代码
根据《人的基因序列变化与人体疾病表征》,CHILD综合征的数据库代码如下:
|
Disease Ontology DOID:0111822
OMIM® 308050
SNOMED-CT 17608003
ICD10 via Orphanet Q87.8
|
UMLS via Orphanet C0265267
Orphanet ORPHA139
MedGen C0265267
SNOMED-CT via HPO 103276001 111266001 11375002 15188001 200766001 20255002
203598005 205456006 237774001 237836003 238810007 239107007 247441003 248409006 249696007 25201003 253273004 254156001 268282005 26996000 271811009 278040002 32958008 343087000 360507004 367524008 385522000 396228006 396347007 396351009 399955009 43064006 444827008 45503006 48334007 52781008 55033002 55726006 56317004 57048009 59494005 64217002 65068000 70819003 708474007 7890003 80281008 80825009 86735004 86765009 88565003 93250003 95828007
UMLS C0265267
|
CHILD综合征基因解码、基因检测有什么用?
选基因解码项目,可以找到发病原因,寻找目前可以进行有效治疗的药物。指导婚恋方式,提供生育健康二胎的建议。
- 【佳学基因检测】代谢性遗传病,基因检测有必要吗?...
- 【佳学基因检测】内分泌和神经内分泌肿瘤基因检测...
- 【佳学基因检测】内分泌瘤临床级基因检测诊断机构...
- 【佳学基因检测】内分泌瘤的基因诊断基因检测...
- 【佳学基因检测】多发性内分泌腺瘤综合征 (MEN)基因检测...
- 【佳学基因检测】遗传性内分泌疾病基因诊断方案...
- 【佳学基因检测】垂体疾病基因检测项目...
- 【佳学基因检测】2024肾上腺疾病基因检测项目...
- 【佳学基因检测】甲状腺疾病基因检测项目大全...
- 【佳学基因检测】内分泌科基因检测项目大全...
- 【佳学基因检测】糖尿病基因检测项目介绍...
- 【佳学基因检测】基因检测如何帮助治疗中链酰基辅酶A脱氢酶缺乏症?...
- 【佳学基因检测】戊二酸血症Ⅰ型遗传性阻断基因检测...
- 【佳学基因检测】PA基因检测及丙酰辅酶A羧化酶缺乏症基因筛查...
- 【佳学基因检测】MMA(甲基丙二酸血症)基因检测实例展示...
- 【佳学基因检测】酪氨酸血症不同亚型的正确诊断案例...
- 【佳学基因检测】江西南昌高酪氨酸血症全外显子基因检测结果判断...
- 【佳学基因检测】瓜氨酸血症基因检测与基因筛查...
- 【佳学基因检测】高甲硫氨酸血症基因检测与基因筛查...
- 【佳学基因检测】CAH基因检测在哪儿做?先天性肾上腺皮质增生症...
- 【佳学基因检测】做无丙型球蛋白血症非布鲁顿型基因解码、基因检测采用什么样品?...
- 【佳学基因检测】做Berardinelli-Seip先天性脂肪营养不良基因解码、基因检测方便吗?...
- 【佳学基因检测】巴一柯综合征Bassen-Kornzweig syndrome基因解码基因检测...
- 【佳学基因检测】安徒生糖原贮积症基因解码、基因检测...
- 【佳学基因检测】甲状腺激素抵抗基因解码、基因检测怎么预约解读?...
- 【佳学基因检测】如何做甲状腺激素抵抗全身性抵抗型基因解码、基因检测?...
- 【佳学基因检测】常染色体显性广泛性甲状腺激素抵抗基因解码、基因检测的报告有人解读吗?...
- 【佳学基因检测】β-半乳糖苷酶-1(GLB1)缺陷基因检测、基因解码...
- 【佳学基因检测】做糖蛋白VI缺乏症基因解码、基因检测需要多少钱?...
- 【佳学基因检测】强迫症基因解码、基因检测的报告看得懂吗?...
- 来了,就说两句!
-
- 最新评论 进入详细评论页>>
