












【佳学基因检测】癫痫基因检测为什要包含CNV的检测?
癫痫基因检测增加检出率的要求:
拷贝数变异 ( CNV ) 是多种神经发育障碍(包括癫痫)的致病基因突变。随着基因解码的应用,更多关于癫痫的致病基因知识的增加,需要更新对癫痫基因测序数据的分析。癫痫基因检测基因检测对癫痫的发生与拷贝数变异的关系进行了分析,以便找到患者的发病原因,并提高检出率。癫痫基因检测汇集了 1255 名患者,这些患者均已存在基于阵列比较基因组杂交或单核苷酸多态性阵列的CNV数据。所有患者均患有“癫痫加”,即癫痫伴有合并症,包括智力障碍、精神症状以及其他神经和非神经系统特征。CNV分类采用适合癫痫的系统过滤工作流程进行。在基因数据质量控制后剩下的 1097 名患者中,120 人(10.9%)携带至少一种被归类为致病的常染色体CNV;19 人(1.7%)携带至少一种被归类为可能致病的常染色体CNV。11名患者(1%)携带不止一种(可能)致病的CNV。癫痫基因检测确定了涵盖最近报告的(HNRNPU)或新出现的(RORB )癫痫基因的CNV,并进一步描述了与这些基因突变相关的表型。癫痫基因检测的癫痫致病基因鉴定分析发现了其他新的癫痫候选基因。将致病性CNV携带者的表型特征与非致病性CNV携带者的表型特征进行比较,癫痫基因检测发现患有非神经系统合并症(尤其是畸形)的患者更有可能携带致病性CNV(风险比 = 4.09,置信区间 = 2.51‐6.68;P = 2.34 × 10 −9)。包括已发表对照组数据的荟萃分析表明,癫痫的存在与否并不影响CNV的检测频率。
使用专门调整的工作流程,可以在 10.9% 的癫痫合并症患者中识别出致病性常染色体CNV ,如果癫痫基因检测同时考虑可能致病的CNV ,这一比例将上升至 12.7% 。癫痫基因检测的数据表明,具有合并症特征的癫痫应被视为选择患者进行包括CNV检测在内的诊断算法的指征。协作性大规模CNV重新分析可在原因不明的病例中重新声明致病性,并可促进有希望的癫痫候选基因的发现。
癫痫基因检测为什么要包含CNV的检测关键词
阵列CGH、拷贝数变异、癫痫基因、SNP阵列
关键点:
- CNV 是导致癫痫的重要因素,近 13% 的病例存在致病性和可能致病性的 CNV
-
使用基因解码分析流程对 CNV 进行分类,可以分析通过不同平台筛选的回顾性收集患者的数据
-
癫痫致病基因鉴定基因解码重点介绍了最近报道的(HNRNPU)或新出现的(RORB)癫痫基因的 CNV,并进一步描述了相关的表型
-
患有非神经系统合并症(尤其是畸形)的患者更有可能携带致病性 CNV
癫痫基因检测为什么需要纳入CNV的检测?
目前的估计表明, 50 % -70 % 的癫痫是由遗传因素引起的。拷贝数变异(CNV) 是导致某些癫痫的主要风险类型。全基因组寡核苷酸阵列 CGH 或 SNP 阵列通常用于评估疑似有遗传原因的复杂表型患者。6 据报道,在患有不同类型癫痫的患者中,约 5%-12% 存在 CNV 风险或病因。2、4、5、7、8、9 一项针对222名患者的癫痫致病基因鉴定分析显示,同时存在智力障碍 ( ID ) 、畸形特征、自闭症谱系障碍( ASD ) 、耐药性或其他合并症会增加致病性 CNV 的风险。10复发性 CNV“热点”会导致不同类型的癫痫。5、11 CNV检测已指向新的癫痫基因。12
需要对癫痫中可能相关的 CNV 的频率和类型进行稳健估计,以确定是否应将 CNV 检测纳入具有各种癫痫表型的患者的遗传评估中。随着对癫痫遗传学的了解不断增加,对遗传数据进行系统、反复的重新评估变得至关重要。这一过程需要收集大量个体,而且由于这些数据必然来自使用不同技术的不同中心,因此必须有一种稳健的联合重新评估方法。
癫痫通常是神经发育障碍 (NDD) 的一个特征。最近一项针对 NDD 和癫痫患者的癫痫致病基因鉴定分析报告称,确诊为癫痫性脑病 (EE) 的个体和确诊为患有未指明癫痫的 NDD 的个体的罕见变异频率结果相似,13这表明,从遗传学上讲,癫痫可以被视为 NDD 谱的一部分。从 CNV 的角度来看待这一概念,并确定特定癫痫表型中 CNV 的频率,癫痫基因检测召集了一大批具有“癫痫加”表型的国际患者,癫痫基因检测将其定义为癫痫和合并症特征的发生,包括 ID 和精神、神经和非神经系统特征。使用基于当前 CNV 分类知识的工作流程系统地癫痫致病基因鉴定分析了预先存在的阵列数据。该工作流程能够结合多中心 CNV 数据,提供对 CNV 对癫痫的贡献的稳健、最新的重新评估,并识别新的候选致病常染色体 CNV。该方法可以在未来的时间点与其他队列迭代应用,从而充分利用现有数据。
参与分析的患者数据
预先存在的 CNV 数据来自为临床或癫痫致病基因鉴定分析目的而进行的阵列 CGH 或 SNP 阵列,这些数据来自 8 家癫痫和/或遗传病专科中心。所有患者还具有合并症特征,包括 ID、自闭症、畸形特征、其他神经或非神经系统疾病、结构性脑异常或多药耐药性。14临床信息由转诊临床医生收集。癫痫发作和癫痫/综合征类型根据国际抗癫痫联盟标准(如有)进行分类。
CNV分析:质量控制和分类
所有 CNV 检出均由癫痫基因检测合作医院。图1显示了癫痫基因检测用于分类 CNV 的工作流程。癫痫基因检测仅关注常染色体 CNV,因为非性染色体的 CNV 调用质量更高。为确保高可靠性,癫痫基因检测仅考虑根据以下标准具有高调用置信度的 CNV:(1)大小 ≥ 150 kb,(2)SNP 阵列的连续探针覆盖率 ≥30 个,阵列 CGH 的探针覆盖率 ≥3 个,和(3)整个癫痫致病基因鉴定分析样本中的微缺失/微重复频率 < 1%。如果样本的总缺失或重复(或两者)调用数与整个数据集中任何调用/样本的平均调用数相差 >2 SD,则将样本排除在分析之外。进一步的人工分析使用了基于当前对分类的理解定制的工作流程,包括美国医学遗传学学院指南19和其他文献。 CNV 被分为四类:致病性、可能致病性、良性或意义不明。简而言之,工作流程如下:首先,健康人群中存在的常见 CNV 20被归类为“良性”。如果其余 CNV满足以下标准,则将其归类为“致病性”:癫痫致病基因鉴定分析 CNV 与已知与癫痫相关的任何 CNV 重叠≥80%;或 CNV 大小≥3 Mb;或 CNV 大小<3 Mb 且>1 Mb,并且是新发的。其余 CNV 根据其基因内容进一步分类。如果 CNV 涉及已知与癫痫相关的基因,其表型与文献中的一致,并且 CNV 类型(缺失/重复)与当前对基因改变(功能获得或丧失)的致病机制的认识相符,则该 CNV 被归类为致病性。如果 CNV 包含与癫痫相关的基因,但不满足其他条件,则只有在证明是从头发生的的情况下才认为该 CNV 是致病的,否则则归类为“可能致病”。根据已发表的数据集21和数据库 GTEx(http://www.gtexportal.org/home/),包含脑表达基因的 CNV 只有在从头发生的情况下才被归类为“可能致病”。由于大多数 CNV 平台在调用纯合缺失或重复方面的限制,因此不考虑癫痫基因的隐性遗传分析。其余 CNV 被归类为“意义不明”。
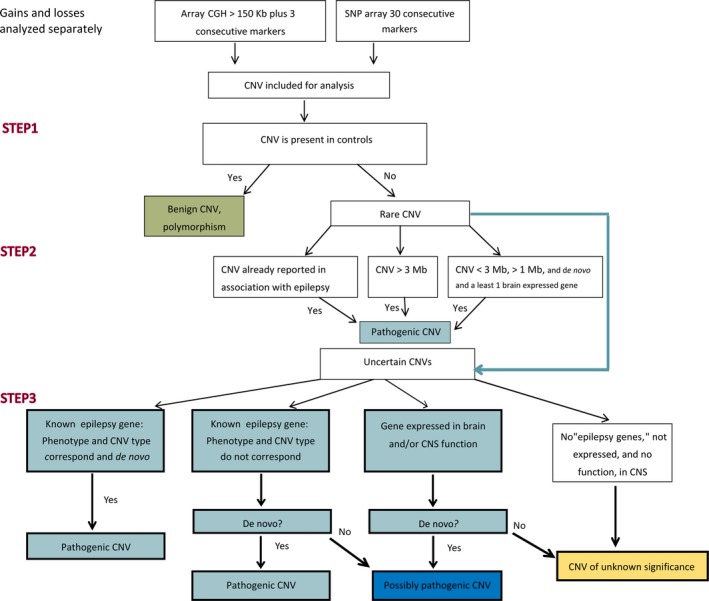
图1:用于分类癫痫基因检测患有癫痫等疾病的患者队列中的拷贝数变异 ( CNV ) 的工作流程。显示了将CNV分类为良性、致病性、可能致病性和意义不明组的分步程序。CGH,比较基因组杂交;CNS,中枢神经系统;SNP,单核苷酸多态性
癫痫基因检测所应包含的致病性CNV
癫痫基因检测汇集了 1255 名患者。经过质量控制后,1097 名患者被保留进行分析。其中,139 名 (12.7%) 携带总共 142 个常染色体 CNV,分类为致病性 (n = 122, 10.9%) 或可能致病性 (n = 20, 1.7%)。11 名患者 (1%) 携带两个致病性或可能致病性的 CNV。
致病性CNV
为了简化介绍,癫痫基因检测进一步将致病性 CNV 分为四个亚组:(1)有充分证据表明在癫痫中富集的复发性 CNV;(2)与遗传性在线孟德尔遗传数据库 (OMIM) 综合征相关的 CNV,该综合征可表现为癫痫的神经系统症状;(3)未知的 CNV 是否会在癫痫中富集,且与任何其他 OMIM 综合征无关,但包含至少一个已经与癫痫有关的基因;(4)基于大小和从头发生的 CNV。
癫痫中反复出现的、有据可查的 CNV
36 例患者患有癫痫,且已知其为复发性 CNV(36/120,30%;表格1)。一名患者有 2 个复发性致病性 CNV。16p13.11 缺失最为常见,在 120 名致病性 CNV 患者中发生 10 名(8.3%),在 1097 名癫痫致病基因鉴定分析对象中发生 10 名(0.9%)。其他常见 CNV 包括 1p36 缺失(OMIM #607872,120 名患者中 5 名,4.2%)、15q11.2 缺失(OMIM #615656,120 名患者中 5 名,4.2%)和 22q11.2 19重复(OMIM #608363,120 名患者中 5 名,4.2%)。
表1:复发性 CNV 在癫痫中具有明显富集
| 样本,n | Chr区域 | CNV类型 | 综合症 | OMIM/参考边界 | OMIM 或参考文献 |
|---|---|---|---|---|---|
| 5 | 1p36 | 删除 | 1p36 染色体缺失综合征 | 1:1‐27 600 000 | #607872 |
| 2 | 1q21.1 | 删除 | 1q21.1 染色体缺失综合征 | 1:143 200 000‐147 500 000 | #612474 |
| 1 | 1q21.1 | 复制 | 1q21.1 染色体重复综合征 | 1:143 200 000‐147 500 000 | #612475 |
| 5 | 15q11.2 | 删除 | 15q11.2 染色体缺失综合征 | 15:20 500 000‐25 500 000 | #615656 |
| 3 | 15q13.3 | 删除 | 15q13.3 染色体缺失综合征 | 15:30 900 000‐33 400 000 | #612001 |
| 3 | 16p11.2 | 删除 | 16p11.2 染色体缺失综合征 | 16:28 500 000‐35 300 000 | #611913 |
| 10 | 16p13.11 | 删除 | 16p13.11 染色体缺失综合征 | 16:15 000 000‐16 300 000 | |
| 3 | 22q11.21 | 删除 | 22q11.2 染色体缺失综合征,远端 | 22:17 40万‐2550万 | #611867 |
| 5 | 22q11.21 | 复制 | 22q11.2 染色体重复综合征 | 22:17 40万‐2550万 | #608363 |
CNV,拷贝数变异;OMIM,在线人类孟德尔遗传数据库。
与遗传性 OMIM 综合征相关的 CNV,该综合征具有以癫痫为特征的神经系统症状
33 名个体具有致病性 CNV(33/120,27.5%),这些 CNV 位于与神经系统特征(包括癫痫)相关的明确遗传综合征的区域,并且与相关综合征相符。最常见的如下:Williams‐Beuren 7q11.23 缺失综合征(5 名患者)、15q11.2 远端重复综合征(3 名患者)、包括PRRT2的 16p11.2 重复综合征(4 名患者)、Potocki‐Lupski 17p11.2 重复综合征(2 名患者)和 17p13.3 缺失综合征,也称为 Miller‐Dieker 无脑畸形缺失综合征(3 名患者)。癫痫基因检测还在 2q24.3 22和 4p16.3-p13 23处发现了从头重复,这些区域的缺失和相互重复都与癫痫有关。
CNV(包括癫痫相关基因)
19 例患者有 CNV(19/120,15.8%),包括癫痫相关基因(表2)。五个人的 CNV 包括HNRNPU(四个新生缺失和一个重复;两个缺失和重复还包含侧翼AKT3基因)。携带缺失的四个先证者表现为癫痫,一名患者被归类为 Lennox‐Gastaut 综合征,另一名患者被归类为遗传性全身性癫痫 (GGE),其余两名患者被归类为未分类的早发性耐药性癫痫。四名患者报告有中度至重度 ID,一名还患有 ASD。三名患者中有一名患有小头畸形,是先天性的和严重的(-4 SD)。脑磁共振成像显示四名患者中有三名出现胼胝体发育不全或发育不全。三名患者出现面部畸形特征。携带大量(>100 Mb)重复的患者,涉及HNRNPU和AKT3等许多其他基因,具有复杂的表型,包括新生儿癫痫发作、多小脑回和多种心脏缺陷。三个人有 9q21.13 缺失,一个是新生缺失,两个是未知遗传的缺失,包括一个最近描述的与癫痫有关的基因RORB。所有患者均表现为 ID 和全身性癫痫,伴有失神或非典型失神,两例有眼睑肌阵挛,一例有光敏性。三个缺失包括ADGRV1基因,其中两个包括MEF2C。表2中列出了在单个患者中发现缺失或重复的其他癫痫基因表22包括GNAO1、NEDD4L和SIK1。
表 2:包括癫痫相关基因CNV
| 病例 | CNV类型 | Chr区域 | 开始 | 停止 | 大小,Mb | 遗产 | 癫痫基因 | 癫痫表型 | 其他临床特征 | 神经影像学 | 已报道的与基因相关的癫痫表型 | 已报道的癫痫基因的拟议疾病机制(功能获得或丧失) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IT_FLO_041 | 删除 | 1q42‐q44 | 236852056 | 249212809 | 12.4 | 从头 | 氢核糖核酸酶3 | 癫痫 NOS、DR | ID、刻板行为、先天性小头畸形(-4 SD)、面部畸形 | CC 发育不全,全前脑畸形 |
HNRNPU:早期婴儿癫痫性脑病,54(MIM 617391) AKT 3:巨脑畸形、多小脑回畸形、多指畸形、脑积水综合征 2(MIM 615937) |
功能丧失(表格 S6a); 功能丧失和获得(表格 S6a了解更多详情) |
| BE_LEU_127 | 复制 | 1q21.1‐q44 | 144967252 | 249212666 | 104.2 | 未知 | 氢核糖核酸酶3 | 婴儿期发病的癫痫 NOS、DR | 肌张力低下、呼吸功能不全、心脏缺陷(升主动脉和主动脉弓较大、博塔利导管开放、ASD2 伴有小左/右分流;肺发育不全)、肾旋转不良、面部畸形 | 侧脑室和脑腔增宽,多小脑回畸形 | ||
| PO_W_031 | 删除 | 1q43‐q44 | 241757184 | 245072885 | 3.3 | 从头 | 氢核糖核酸酶3 | 来源不明的焦点 | ID、肌张力低下、获得性小头畸形(-2 SD)、面部畸形、肌张力低下 | 额叶萎缩和 CC 发育不全 | ||
| IT_FLO_062 | 删除 | 1q44 | 244515959 | 247118959 | 2.6 | 从头 | 氢核聚变 | 全身性癫痫,DR | ID、面部畸形、GH 缺陷、耳聋、后天性小头畸形(-2 SD)、关节过度松弛、脊柱侧弯 | CC发育不全,心室不对称 | ||
| BE_LEU_009 | 删除 | 1q44 | 244823848 | 248093878 | 3.3 | 从头 | 氢核聚变 | Lennox‐Gastaut 综合征 | ID、脊柱侧弯、胃食管反流、双侧角膜混浊 | 髓鞘形成延迟、透明隔萎缩、导水管狭窄、脑积水 | ||
| BE_ANT_005 | 删除 | 2q24.3 | 163860225 | 172528095 | 8.7 | 从头 | SCN1A、SCN2A | 不明原因的全身性癫痫 | 智力缺陷、面部畸形 | 消极的 |
SCN1A:早期婴儿癫痫性脑病,6(Dravet 综合征;MIM 607208);全身性癫痫,伴有热性惊厥附加症,2 型(MIM 604403);家族性热性惊厥,3A(MIM 604403); SCN2A:早期婴儿癫痫性脑病,11(MIM 61372);良性家族性婴儿癫痫,3(MIM 6077451) |
功能丧失;功能丧失与 ASD 相关,功能获得与 EE 相关 |
| IT_FLO_020 | 删除 | 5q14.3 | 88232244 | 90181244 | 1.9 | 从头 | ADGRV1 | 癫痫和 FS NOS | 没有任何 | 异常 NOS |
ADGRV1:家族性热性惊厥,4(MIM 604352);肌阵挛性癫痫(表 S2); MEF2C:智力低下、刻板运动、癫痫和/或脑畸形(MIM 613443) |
功能丧失 |
| PO_W_027 | 删除 | 5q14.3‐q15 | 87100153 | 92514871 | 5.4 | 从头 | ADGRV1,MEF2C | 癫痫(非特指特殊类型) | 智力缺陷、畸形 | 不适用 | ||
| IT_FLO_024 | 删除 | 5q14q21 | 87770000 | 95780000 | 8 | 未知 | ADGRV1,MEF2C | 癫痫(非特指特殊类型) | ID、大头畸形、面部畸形 | 脑室周围结节性异位 | ||
| BE_LEU_211 | 删除 | 5q34 | 161059999 | 161446505 | 0.4 | 未知 | GABRA1、GABRA6 | 癫痫(非特指特殊类型) | ID | Corticosubcortical atrophy, supratentorial ventricular enlargement, periventricular vascular leukoencephalopathy, white matter lesions, lacunar infarcts in the basal ganglia and left thalamus |
GABRA1: epileptic encephalopathy, early infantile, 19 (MIM 615744); possible susceptibility allele; juvenile myoclonic epilepsy (MIM 611136) and childhood absence epilepsy (MIM 611136); GABRA6: possible susceptibility allele for childhood absence epilepsy (Table S2) |
Loss of function |
| PO_W_019 | Deletion | 9q21.13 | 74741400 | 77306932 | 2.6 | De novo | RORB | Generalized photosensitive epilepsy (Jeavons syndrome) | ID, autism, strabismus | Negative | Generalized epilepsy, ID (Table S6b for more details) | Loss of function |
| BE_LEU_244 | Deletion | 9q21.13 | 76474486 | 81651005 | 5.2 | Unknown | RORB | Generalized of unknown origin | ID, episodic ataxia | Small nonspecific white matter lesions over right parietal hemisphere | ||
| US_267 | Deletion | 9q21.12‐q21.13 | 72702925 | 77128468 | 4.4 | Unknown | RORB | Generalized epilepsy of unknown origin | ID, pyramidal sign, tremor, neurogenic bladder, psychotic episodes, severe macrocytic anemia, cold agglutinin disease, bilateral femuropatellar arthrosis, facial dysmorphisms | NA | ||
| BE_LEU_205 | Deletion | 12p13.31 | 8691730 | 14215925 | 5.5 | Unknown | GRIN2B | Focal epilepsy of unknown origin | ID, facial dysmorphism | Negative | Epileptic encephalopathy, early infantile, 27 (MIM 616139) | Loss and gain of function |
| PO_W_017 | Duplication | 14q11.2‐q12 | 23309096 | 31675172 | 8.3 | De novo | FOXG1 | Epilepsy NOS | ID | NA | Rett syndrome, congenital variant (MIM 613454) | Loss of function |
| IT_FLO_033 | Deletion | 16q12.1‐q21 | 52347499 | 64578499 | 12.2 | Unknown | GNAO1, GPR56 | Generalized epilepsy of structural origin | ID, language delay, facial dysmorphism, microcephaly, cryptorchidism | Polymicrogyria | GNAO1: epileptic encephalopathy, early infantile, 17 (MIM 615473); neurodevelopmental disorder with involuntary movements (MIM 617493); movement disorder with or without EE; GPR56: polymicrogyria (MIM 606854, 615752) |
Loss of function; gain of function (recessive, loss of function) |
| IT_FLO_017 | Deletion | 18q21.31‐q21.33 | 54687002 | 59222020 | 4.5 | Unknown | NEDD4L | Focal epilepsy of unknown origin | ID, hypotonia, dyspraxia, clumsiness, convergent strabismus | Vermis hypoplasia | OMIM: periventricular nodular heterotopia (MIM 617201; Lennox‐Gastaut syndrome–infantile spasms) | Loss of function |
| IT_FLO_074 | Deletion | 20q13.33 | 61845191 | 62893189 | 1.1 | De novo | KCNQ2, CHRNA4 | Generalized epilepsy of structural origin | Bilateral deafness, facial dysmorphism, lumbar kyphosis, sacral dimple, bilateral clinodactyly, small hands and fingers, hypoplastic flexion creases, atrial and ventricular septal defects, left renal agenesis defects, left renal agenesis | Periventricular nodular heterotopia |
KCNQ2: epileptic encephalopathy, early infantile, 7 (MIM 613720); myokymia (MIM 121200); seizures, benign neonatal, 1 (MIM 121200); CHRNA4: epilepsy, nocturnal frontal lobe, 1 (MIM 600513) |
Gain and loss of function; loss and gain of function |
| US_073 | Deletion | 21q22.3 | 43420839 | 46944323 | 3.5 | Unknown | SIK1 | Generalized epilepsy of unknown origin | ID, ataxia, spasticity, kyphoscoliosis, aortic valve deficiency | 侧脑室扩大,伴有枕角突出(空洞头畸形) | 早期婴儿癫痫性脑病,30(MIM 616341) | 功能丧失 |
与每个已知癫痫基因相关的报告表型是指 OMIM 中报告的表型,如果没有,则指补充材料中注明的引用。
ASD,房间隔缺损;CC,胼胝体;CNV,拷贝数变异;DR,耐药性;EE,癫痫性脑病;FS,热性惊厥;GH,生长激素;ID,智力障碍;MIM,人类孟德尔遗传;NA,不可用;NOS,未另行指定;OMIM,人类孟德尔遗传在线数据库。
基于大小和新生发生的致病性常染色体 CNV
32 个人(32/120,26.6%)的 CNV 仅属于这一类(一个个体有两个大的致病性 CNV)。癫痫基因检测在健康个体中未发现重叠的 CNV(基因组变异数据库;http: //dgv.tcag.ca/dgv/app/home )。在表现出 ID、癫痫和畸形等各种临床特征的患者中,16 个(16/32,50%)≥ 3 Mb 的 CNV 与 Decipher(https://decipher.sanger.ac.uk/)中描述的 CNV 重叠或部分重叠(表 S4b )。有趣的是,在一名 EE 患者中,癫痫基因检测发现了包括NBEA在内的新发 13q33.1-q13.3 缺失。
可能致病的CNV
19 个人(19/1097,1.73%)共有 20 个 CNV 被归类为可能致病(1 个人有两个可能致病的 CNV);10 个是新生的(表3)。19 人中有 17 人(20 个 CNV 中有 18 个)的 DNA 可用来使用 MAQ 分析检查 CNV 和/或遗传。在分析的 18 个 CNV 中,有 11 个得到确认;在 7 个案例中,测试结果不确定(表3)这些 CNV 被归类为可能致病,因为它们包含癫痫基因,但是是遗传的,或者变化的方向与已知的疾病机制(功能的丧失或获得)或表型不一致,或者因为它们包含脑表达基因并且是新发的。属于第一类的 CNV 是母系遗传的 10q23 缺失,包括LGI1,以及母系遗传的 20q13 重复,包括KCNQ2、CHRNA4和EEF1A2。另外四个遗传的 CNV 包括隐性基因:PLCB1、TBC1D24、ABAT和CNTNAP2。不能排除另一个等位基因上可能存在的额外单核苷酸变异 (SNV)。值得注意的是,PLCB1缺失被证实是纯合的,将被认为是致病的,但癫痫基因检测的流程图并非针对隐性分析而开发。
表3:常染色体 CNV 被归类为“可能致病”
| 病例 | CNV类型 | Chr区域 | 开始 | 停止 | 大小,Mb | 遗产 | 建议候选基因 | 癫痫表型 | 其他临床特征 | 神经影像学 | MAQ 验证 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BE_LEU_009 | 复制 | 1q43 | 239842929 | 240356854 | 0.5 | 从头 | FMN2(启动基因内的BP)、CHRM3(停止基因内的BP) | 癫痫(非特指特殊类型) | ID、脊柱侧弯、胃食管反流、双侧角膜混浊 | 髓鞘形成延迟、透明隔萎缩、导水管狭窄、脑积水 | 从头 |
| US_184 | 复制 | 3q28 | 191886383 | 192432844 | 0.5 | 从头 | FGF12(基因内重复) | 癫痫(非特指特殊类型) | 学习障碍、注意力缺陷 | 左海马前部和中部旋转不良 | 不适用 |
| BE_LEU_141 | 删除 | 3q22.3 | 136035522 | 136412948 | 0.4 | 从头 | STAG1、PCCB(基因内起始BP) | 癫痫(非特指特殊类型) | 智力残疾、自闭症、肌张力亢进、脊柱侧弯、 | 不适用 | 患者确诊,母亲未确诊 |
| IT_FLO_036 b | 复制 | 4q21.22‐q21.23 | 84035965 | 84813544 | 0.8 | 从头 | COQ2(阻止基因内的 BP) | 肌阵挛性失张力癫痫 | ID | 消极的 | 从头 |
| IT_FLO_127 | 删除 | 5q23.2 | 122481284 | 122987185 | 0.5 | 从头 | CEP120、CSNK1G3(基因内停止BP) | 肌阵挛性癫痫 | ID,肌张力低下 | 消极的 | 从头 |
| PO_W_039 | 复制 | 7q35‐q36.1 | 146934489 | 148471787 | 1.5 | 遗传(男) | CNTNAP2(基因内的起始BP) | 癫痫(非特指特殊类型) | ID | CC发育不全 | 尚无定论 |
| IT_FL0_131 | 删除 | 6q26 | 161725639 | 161878527 | 0.2 | 从头 | PARK2(基因内停止BP) | 癫痫 NOS、FS | 智力低下、肌张力低下、肥胖、双手双脚手指拥挤、甲营养不良 | 不适用 | 尚无定论 |
| 删除 | 12p12.3 | 15469971 | 16375910 | 0.9 | 从头 | 带子 | |||||
| IT_FLO_109 | 复制 | 8p23.3‐p23.2 | 161272 | 801514 | 0.6 | 平衡易位的不平衡分离(M) | FBXO25 | 不明原因的全身性癫痫,DR | 语言障碍 | 消极的 | 尚无定论 |
| IT_FLO_134 | 删除 | 8p23.3‐23.2 | 221611 | 801373 | 0.6 | 平衡易位的不平衡分离(M) | FBXO25 | 肌阵挛性癫痫 | 不 | 不适用 | 尚无定论 |
| BE_LEU_236 | 复制 | 9q22.31 | 95208377 | 95590171 | 0.4 | 从头 | BICD2 | 癫痫(非特指特殊类型) | ID | 不适用 | 从头 |
| IT_FLO_144 | 删除 | 10q23.33 | 95490322 | 95791986 | 0.3 | 遗传(男) | 地表水1 | 癫痫性脑病 NOS | 严重智力残疾、四肢瘫痪、先天性心肌病(植入起搏器) | 脑萎缩小头畸形 | 母系遗传 |
| BE_LEU_012 | 复制 | 15q13.2 | 32509932 | 1.6 | 遗传(P) | CHRNA7 | 来源不明的焦点 | ID | 不 | 尚无定论 | |
| 英国_L_056 | 复制 | 16p13.3 | 2481289 | 2888632 | 0.4 | 未知 | TBC1D24 | 肌阵挛性失张力癫痫 | ID | 消极的 | 先证者确诊,父母无 |
| US_175 | 删除 | 16p13.2 | 8368145 | 8860296 | 0.5 | 遗传(男) | ABAT(基因内停止 BP) | 癫痫性脑病 NOS | 智力低下、失用症、运动障碍、全身肌张力低下 | 消极的 | 母系遗传 |
| IT_FLO_023 | 删除 | 17q21.31 | 43160474 | 43922220 | 0.8 | 从头 | NMT1(基因内起始BP)、PLCD3(基因内起始BP) | 原因不明的局灶性癫痫 | ID、大头畸形、面部畸形、心脏缺陷、皮肤色素异常 | CC发育不全 | 尚无定论 |
| IT_FLO_083 | 删除 | 18q12.3 | 42605437 | 42784321 | 0.2 | 从头 | SETBP1(基因内起始 BP) | 不明原因的全身性癫痫,DR | ID | 消极的 | 从头 |
| BE_LEU_116 | 复制 | 19p13.3 | 538568 | 2268870 | 1.7 | 未知 | 氢核2 | 癫痫 NOS 和 FS | 学习障碍、注意力缺陷多动症、面部畸形 | 消极的 | 先证者确诊,父母无 |
| US_124 | 删除 | 20p12.3 | 8314301 | 8688028 | 0.4 | 继承(M+P) | PLCB1(基因内的起始BP) | 癫痫性脑病 NOS | 严重智力缺陷、小头畸形、肌张力亢进、左侧反射亢进更为明显、左眼斜视 | CT 脑萎缩 | 不适用 |
| 订单号:PO_W_030 | 复制 | 20q13.33 | 61925286 | 62724437 | 0.8 | 遗传(男) | KCNQ2、CHRNA4、EEF1A2 | 原因不明的局灶性癫痫 | 智力缺陷、面部畸形 | 不适用 | 先证者确诊,父母无 |
ADHD = 注意力缺陷多动障碍;BP = 断点;CC = 胼胝体;CNV = 拷贝数变异;CT = 计算机断层扫描;DR = 耐药;FS = 热性惊厥;ID = 智力障碍;M = 母系;MAQ = 多重扩增子定量;NA = 不可用;NOS = 未另行指定;P = 父系。
候选基因列还包括包含已知癫痫基因的 CNV,这些基因由于各种原因尚未被视为致病;例如,变化的方向或表型与文献中报道的不符,或者 CNV 是从受影响状态未知的父母那里遗传的。
在第二类中,癫痫基因检测确定了几个有趣的候选基因,包括一个包括STAG1 的新生缺失和FGF12的新生基因内重复。在这两个基因中,最近才有报道称在患有癫痫等神经发育障碍的患者中存在致病性 SNV。癫痫基因检测进一步确定了与 Shinzel‐Gieidon 综合征和 ID(OMIM 611060 )相关的SETBP1等缺失,以及一个包括HCN2 的重复,HCN2 是一个基因,其中 SNV 发挥功能获得效应,最近被认为是遗传性全身性癫痫的风险因素。HCN2以前也与热性癫痫综合征有关;有趣的是,携带这种 CNV 的患者也有热性惊厥史。他有趣的候选基因位于已识别的从头缺失中或可能被已识别的重复的基因内断点破坏,包括FMN2(也与 AR 智力障碍 MIM616193 相关)、CHRM3、CSNK1G3和NMT1 ,根据最新的 gnomAD( http://gnomad.broadinstitute.org/about)约束指标(https://www.nature.com/articles/nature19057 ) ,它们均在大脑中表达并预测不能耐受功能丧失(功能丧失不耐受的概率 ≥ 0.99)。
富集分析
癫痫基因检测收集了七种特征的表型信息(只要这些信息可用):神经或精神疾病(528/956 例患者,55.2%)、ID(727/944 例患者,77%)、合并非神经系统疾病(242/882 例患者,27.4%)、面部畸形(209/769 例患者,27.2%)、脑异常(288/613 例患者,47%)、1 岁前癫痫发作(175/340 例患者,51.5%)和已知 EE 综合征的诊断(238/487 例患者,49%)。癫痫基因检测将致病性常染色体 CNV 的携带者与未携带致病性常染色体 CNV 的人进行了比较。致病性 CNV 患者的非神经系统疾病(2.68 倍)和畸形(4.09 倍;图2)。除了整体致病性 CNV 富集外,对大型致病性 CNV(>1 Mb)的检测分别显示出更深层次和更显著的倍数富集,分别为非神经系统疾病和畸形的共病分别为 2.82 和 4.94(图2)。

图 2:富集分析。左图:在癫痫致病基因鉴定基因解码中分析的所有患者中,致病性拷贝数变异 ( CNV ) 患者因同时患有非神经系统疾病或畸形而显著富集。右图:将分析限制在携带大型致病性CNV (>1 Mb) 的患者。这些CNV携带者因非神经系统疾病和畸形而特别富集。三角形表示多重检验校正后显著的优势比 (OR)。
基因检测大数据分析
搜索发现 4806 篇引文,其中 59 篇论文符合纳入标准并被纳入系统评价。总体荟萃分析表明,在患有 ID 但不伴有癫痫的患者中,致病性 CNV 的发生率为 15%(95% 置信区间 [CI] = 14-17),在患有精神/神经系统疾病但不伴有癫痫的患者中,发生率为 8%(95% CI = 5-12;图 S3,表4)。这些数据与癫痫基因检测队列中的两个亚组进行了比较:(1)患有癫痫和智力障碍(包括自闭症特征)的患者,产率为 13.5%(95% CI = 9.2‐18.9);(2)患有癫痫和精神/神经系统合并症的患者,产率为 10%(95% CI = 7.9‐11.7)。癫痫基因检测没有发现这些比较中的任何一个具有统计学上的显著差异(异质性检验的P值 >0.05)。
表4:通过荟萃分析比较癫痫致病基因鉴定基因解码中三组患者与文献中三组患者的致病拷贝数变异率
| 癫痫致病基因鉴定基因解码的成果 | 荟萃分析的成果 | 异质性检验的P | |||
|---|---|---|---|---|---|
| 表型(患者,n) | 产率,% (95% 可信区间) | 表型 | 产率,% (95% 可信区间) | ||
| A | 智力残疾+癫痫(207) | 28/207 = 13.5% (9.2‐18.9) | ID | 15%(14-17岁) | 0.4491 |
| b | 精神/神经系统合并症+癫痫(528) | 53/528=10.0%(7.9-11.7) | 精神/神经疾病 | 8%(5-12岁) | 0.3962 |
| C | 癫痫‐EE (238) | 17/238=7.1%(4.2-11.2) | 非EE型癫痫 | 11%(8-14) | 0.1251 |
CI,置信区间;EE,癫痫性脑病;ID,智力障碍。
癫痫基因检测队列中癫痫-EE 患者的致病性 CNV 产量 (7.1%, 95% CI = 4.2-11.2) 低于荟萃分析中癫痫-非EE 患者 (11%, 95% CI = 8-14; 表4)。
癫痫基因解码基因检测将CNV纳入分析内容
大多数癫痫,特别是在婴儿和儿童时期开始的癫痫,都有显著的遗传因素。近年来,大量的下一代测序、全外显子组测序和全基因组测序癫痫致病基因鉴定分析已经发表,揭示了许多癫痫和癫痫综合征中的单基因突变。然而,CNV 对癫痫,特别是并发合并症的癫痫的贡献还未得到充分癫痫致病基因鉴定分析。大多数已发表的报告都是单中心癫痫致病基因鉴定分析。最大的样本量为 2454 例患者,其中包括 1366 例遗传性全身性癫痫患者,此外还有 281 例罗兰氏癫痫患者和 807 例成人局灶性癫痫患者;最大的专门针对癫痫加表型的队列癫痫致病基因鉴定分析了 222 人。这些系列中报告的致病性CNV 的最大频率为 12%,范围为 5%-12%。这些癫痫致病基因鉴定分析往往侧重于测试时还是儿童的个体。2、5、9罕见CNV在患有神经精神疾病(包括原因不明的智力残疾、先天性异常和癫痫)的患者中的重要性已得到充分认可。因此,临床遗传学家、儿科神经病学家和癫痫病学家通常要求进行染色体阵列 CGH 来对具有此类临床特征的患者进行基因诊断。
然而,在健康对照个体中也可能见到 CNV,而确定新发现的 CNV 的致病性可能具有挑战性。为了评估致病性 CNV 的作用并确定可能的候选基因,癫痫基因检测调查了迄今为止报告的最大的队列中癫痫合并症中 CNV 的发生情况。数据来自8个中心,包括成人和儿童。使用专门针对癫痫的系统过滤程序进行常染色体 CNV 分类。该工作流程是识别、重新分析和重新解释这一回顾性收集队列中 CNV 的重要工具,其中 CNV 测试使用不同实验室的不同平台进行。大约 11% 的癫痫合并症患者携带致病性常染色体 CNV。如果癫痫基因检测还考虑可能致病的 CNV,这个数字达到 12.7%。之前类似的癫痫致病基因鉴定分析报告诊断产量从 ~5% 到 12% 不等。因此,癫痫基因检测的结果符合该范围的上限,这可能主要归因于癫痫基因检测队列的“癫痫加”表型以及应用了标准化工作流程。之前发表的癫痫致病基因鉴定分析报告了类似的致病性CNV产量(分别为9.3 %、8.1% 和 12%),也癫痫致病基因鉴定分析了包括 ID 在内的复杂癫痫患者。总体而言,癫痫基因检测癫痫致病基因鉴定分析和之前类似癫痫致病基因鉴定分析的结果表明,在神经发育障碍的复杂表型中,当癫痫发作与 ID 或其他神经系统和非神经系统合并症相关时,识别出致病性 CNV 的概率高于单独的癫痫。
在应用癫痫基因检测在此建议的工作流程方法之前和之后,癫痫基因检测检查了致病性和可能致病性 CNV 的原始分类(如果可用)(138/142)。癫痫基因检测发现 7.2%(10/138)的病例存在差异。主要变化方向是从最初归类为“意义不明”的 CNV(8/10 CNV)变为“致病性”和“可能致病性”(表 S7)。这是意料之中的,因为有关与癫痫相关的脑表达基因或基因区域的信息有所增加。癫痫基因检测已经确认,随着时间的推移,对现有数据的重新分析至关重要。
癫痫基因检测的癫痫致病基因鉴定分析证实了特定 CNV 在癫痫中的重要性,并拓宽了一些相关的表型谱。
据报道,1q21.1、15q11.2、15q13.3、16p13.11 和 22q11.21 处的复发性微缺失是 GGE 和局灶性癫痫的危险因素。在癫痫基因检测的队列中发现的最常见的 CNV 是 16p13.11 缺失,占致病性 CNV 的 8.3%,支持了与临床环境的明显相关性。
癫痫基因检测还发现了几个 CNV,包括基因HNRNPU (1q44) 和RORB ( 9p21.13),这两个基因最近都与癫痫有关。1q43q44关键区域的微缺失与 ID、畸形、胼胝体异常和癫痫有关。该关键区域包括HNRNPU,它是最相关的候选癫痫基因。最近在患有 ID 和癫痫的个体中发现了HNRNPU中的大约 30 个点突变,主要包括截短、剪接位点和一些错义变异。在癫痫基因检测的队列中,有五名患者携带定位到 1q43q44 关键区域的 CNV,除了HNRNPU基因(其中两次重复和一次缺失)外,染色体重排还包括AKT3基因,这可能导致这些患者出现脑部异常。缺失的患者表现出畸形特征、早发性精神运动延迟和早发性癫痫。这些数据证实了HNRNPU在神经发育和癫痫发生中的作用。
RORB突变最早出现在一名轻度智力障碍和部分性癫痫患者身上。最近,在患有神经发育障碍且大多为 GGE(包括失神发作)的患者中也发现了其他突变。在癫痫基因检测的队列中,三名患者携带包括RORB在内的缺失,并表现出智力障碍和全身性癫痫,包括伴有眼睑肌阵挛的失神发作,一名患者有自闭症特征,这支持了RORB在 GGE 以及更广泛地说在几种神经发育障碍中的作用。
在综合征致病性常染色体 CNV 中,癫痫基因检测发现了三名患者的重复位于 17p11.2 Potocki‐Lupski 综合征区域,该区域与经常出现癫痫的 Smith‐Magenis 缺失综合征相对应。这三名患者的表型与 Potocki‐Lupski 综合征一致;癫痫的发生支持了先前的证据,即癫痫是 17p11.2 重复的一个罕见特征。有趣的是,癫痫基因检测发现了五名患者存在包含Williams‐Beuren 区域的 7q11.23 缺失;其中四人患有 Lennox‐Gastaut 综合征,第五人(之前由 Ramocki 等人34报告)患有全身性耐药性癫痫。
仅因尺寸较大而被归类为致病性的 CNV(表 S4b)占所有致病性 CNV 的 27%(33/122)。这些 CNV 包含大量基因,但受影响个体的表型复杂,癫痫基因检测无法确定与已知遗传综合征或候选癫痫基因的关联。然而,对于一名患有 EE 和大量新生 13q13.1-q13.3 缺失的个体,癫痫基因检测可以建议一个关键基因是NBEA,它通过计算机优先排序方法被报告为可能的 EE 基因35,并且最近与癫痫神经发育疾病有关。
癫痫基因检测归类为大型致病性 CNV 中有四个是遗传的。有趣的是,12q21.31 上的重复是从有自闭症家族史的母亲那里遗传的。Decipher 报告称,一名携带重叠重复的患者患有自闭症。根据癫痫基因检测的算法,癫痫基因检测认为这些 CNV 是致病性的,因为不完全渗透性通常是神经系统疾病和癫痫症的特征,而且癫痫基因检测无法排除遗传父母的相关神经系统特征。
1.7% 的患者被确诊为可能致病的常染色体 CNV。除了结果部分讨论的一些已知癫痫基因之外,癫痫基因检测还提出这些区域中的其他基因可被视为导致癫痫的潜在候选基因,但需要进一步验证。癫痫基因检测发现一个从头 18q12.3 缺失,仅包含SETBP1基因。SETBP1的杂合错义突变导致 Schinzel‐Giedion 综合征 (OMIM #269150),其特征是严重的 ID 和特定的颅面特征,其中也会发生癫痫。导致单倍体不足的突变(例如癫痫基因检测患者的缺失)据报道与一种独特的神经系统综合征有关,该综合征包括轻度至中度 ID,但没有典型的综合征性颅面特征。癫痫致病基因鉴定基因解码中的患者仅表现出严重的癫痫和智力障碍,这表明SETBP1突变表型可能比之前描述的更广泛。一名患者有微缺失,在此归类为可能致病,其中包括STAG1,目前与癫痫有关,是一种黏连蛋白病,一名患者携带FGF12的新生基因内重复,其中 SNV 最近在患有癫痫性脑病的患者中被报道。
表3中突出显示了其他有趣的候选基因,包括HCN2、FMN2、CHRM3、CSNK1G3和NMT1。
癫痫基因检测癫痫致病基因鉴定分析队列中的 11 名患者(1%)患有双重打击(包括致病性和可能致病性的 CNV)。在这里,CNV 负担本身就可能导致神经发育表型;正如 Girirajan 及其同事所表明的那样,42 名患有两个或两个以上意义不明的罕见和大型 CNV 的儿童与对照组相比,发育迟缓的可能性高出八倍,这可能是由于剂量敏感基因的破坏所致。然而,癫痫基因检测注意到,癫痫基因检测的分析仅关注具有某种致病意义的 CNV,因此无法深入了解每个患者的 CNV 总体负担。
富集分析显示,致病性常染色体 CNV 与非神经系统疾病和畸形显著相关(对于大型致病性 CNV 和任何致病性 CNV 均如此);大型致病性 CNV 仅与畸形和非神经系统疾病具有更深远和更显著的关联。先前的癫痫致病基因鉴定分析已经观察到畸形患者的 CNV 富集,4强调了对癫痫和相关合并症患者进行 CNV 检测的重要性。同样,与历史对照相比,癫痫基因检测的数据结果来自系统的文献综述和荟萃分析,证实当表型包括或排除癫痫时,致病性 CNV 的百分比没有显着差异。因此,尽管在患有癫痫的人群中寻找 CNV 无疑是值得的,但这种 CNV 可能不仅仅会导致癫痫,但对于通过癫痫确诊的患者,其他特征的存在表明找到潜在致病性 CNV 的可能性较高。癫痫基因检测假设,尽管癫痫作为一种表型不会对诊断结果产生定量贡献,但它的存在可能与潜在致病性 CNV 的类型、位置和基因内容有关。癫痫基因检测的数据比较了癫痫-EE 患者与癫痫-非EE 历史对照,结果表明 EE 患者的致病性 CNV 产量较低,但差异不显著,这提出了一个可能的假设:当癫痫表现为 EE 时,发现致病性 CNV 的可能性会降低,并且 EE 通常是单基因突变的结果。
除了回顾性结构之外,癫痫基因检测的癫痫致病基因鉴定分析还有局限性。所使用的过滤工作流程使癫痫基因检测能够对所检查的大量 CNV 进行系统分类,但癫痫基因检测认识到它并不完美,可能无法准确地将 CNV 映射到高变染色体区域。由于过滤掉小的 CNV、错误分类异常或与癫痫相关的基因列表不完整(新的癫痫相关基因不断被报道),可能遗漏致病性 CNV。癫痫基因检测将性染色体排除在 CNV 调用和后续分析之外,因为从这些染色体调用拷贝数容易出现假阳性调用,并且可能会夸大诊断相关 CNV 的报告频率,因为 X 染色体尤其与神经发育障碍有关。此外,除非使用另一种方法癫痫致病基因鉴定分析第二个等位基因,否则不能通过这种类型的分析排除隐性疾病。
总之,癫痫基因检测强调了常染色体 CNV 在近 11% 的患者(如果也考虑可能的致病性则高达 12.7%)中具有致病作用,这些患者患有原因不明的癫痫并伴有合并症,并重申应在癫痫患者中寻找 CNV 的概念,尤其是当癫痫合并其他神经系统和非神经系统疾病时。
癫痫致病基因鉴定基因解码为更好地理解和评估在患有癫痫的患者中发现的 CNV 开辟了新的视角。癫痫基因检测表明,使用经过调整的工作流程重新解释预先存在的数据可以突出新的发现,癫痫基因检测建议随着新方法和数据的出现,定期系统地审查预先获得的遗传数据。这里使用的工作流程是专门为癫痫设计的,可用于对通常在不同时间收集的不同队列的数据进行同质化。确定某些 CNV 的致病作用可能具有挑战性,尤其是当 CNV 与已知综合征无关,或者相似的 CNV 可能大小不同、可能包含不同的基因,并且没有可用于帮助解释的家族分离数据时。定制的疾病特异性算法可能有助于将 CNV 分配到比“可能致病”或“意义不明”更明确的诊断类别。仍有一些 CNV 的作用只有通过增加癫痫致病基因鉴定分析病例数量、功能癫痫致病基因鉴定分析以及临床医生和实验室科学家之间的持续交流才能得到澄清。该癫痫致病基因鉴定分析是新成立且不断发展的癫痫 CNV 国际联盟(EpiCNV)的第一个项目,其中大规模数据聚合和共享将被用作癫痫 CNV 和基因识别的新工具。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
