












【佳学基因检测】脊髓小脑共济失调(小脑萎缩)基因检测
脊髓小脑共济失调(小脑萎缩)基因检测导读:
脊髓小脑共济失调(SCA)是指一组在表型和遗传上都存在异质性的常染色体显性脊髓小脑共济失调症。在表型上,它们表现为步态共济失调,经常伴有构音障碍和眼球运动问题。其他体征和症状也很常见,包括各种锥体和锥体外系体征以及智力障碍。脊髓小脑共济失调(SCA) 的遗传原因是疾病基因内的重复扩增或常见突变(点突变、缺失、插入等)。这两种类型的突变通常会导致难以区分的表型(位点异质性)。《脊髓小脑共济失调(小脑萎缩)基因检测》重点介绍由常见突变引起的脊髓小脑共济失调(SCA)。它描述了《人体神经系统疾病及其产生的基因原因》中已经列出的 27 种类型的表型和基因型,并讨论了已鉴定出疾病基因的 21 种类型的分子发病机制。除了主要类型外,《脊髓小脑共济失调(小脑萎缩)基因检测》还总结了由线粒体基因突变引起的脊髓小脑共济失调(小脑萎缩)。根据所描述的各种脊髓小脑共济失调(SCA) 中的发现来考虑需要进行的基因检测,并根据基因检测结果揭示的疾病机制安排设计正确的治疗药物。
脊髓小脑共济失调(小脑萎缩)基因检测关键词
脊髓小脑性共济失调,常见突变,Ca 2+稳态,疾病机制
为什么要花费人力、物力进行脊髓小脑共济失调(小脑萎缩)基因检测
脊髓小脑性共济失调 (脊髓小脑共济失调(SCA)) 是一组常染色体显性共济失调,其特征是小脑退化,常与脑干萎缩相结合,有时也被称为小脑萎缩基因检测、脊髓小脑脑萎缩基因检测。主要临床症状是步态共济失调,通常与构音障碍和视力问题有关。还可能出现许多其他体征和症状,包括认知障碍、肢体和躯干共济失调、震颤、僵硬、运动迟缓、肌张力障碍和反射亢进,并且可能是少数类型的脊髓小脑共济失调(SCA) 的特征。然而,大多数脊髓小脑共济失调(SCA) 无法通过临床进行区分,需要进行基因鉴别。
目前,大约有 50 种不同的脊髓小脑共济失调(SCA),它们通过疾病基因或染色体位置(如果疾病基因尚不清楚)来区分。脊髓小脑共济失调(SCA)疾病基因/位点存在于大多数染色体上。导致疾病的突变通常是疾病基因内串联重复的扩增。这些重复大多由三核苷酸组成(主要是 CAG;有时是 CTA/CTG),但五核苷酸和六核苷酸重复扩增也会导致脊髓小脑共济失调(SCA),例如脊髓小脑共济失调(SCA) 10、37 和 36型。除了重复扩增外,更常见的突变类型(点突变、缺失、插入、重复)是许多不同类型的脊髓小脑共济失调(SCA) 的基础。
《脊髓小脑共济失调(小脑萎缩)基因检测》回顾了特定脊髓小脑共济失调(SCA) 中由非重复扩增突变构成的致病基因突变。描述了每种脊髓小脑共济失调(SCA) 的表型,然后对疾病基因进行了定位和鉴定。给出了疾病基因的正常功能,并讨论了疾病的潜在病理机制。这是致病基因鉴定基因解码的特征,它比以数据库比对为基础的基因检测更接近疾病发生的真相和病理学,对药物设计和遗传阻断更具有指导作用。还将介绍尚未鉴定疾病基因的少数脊髓小脑共济失调(SCA) 类型。《脊髓小脑共济失调(小脑萎缩)基因检测》包括 21 种由常见突变引起的脊髓小脑共济失调(SCA)。在描述不同类型的脊髓小脑共济失调(SCA) 之后,讨论了这些脊髓小脑共济失调(SCA) 的共同特征、基因产物的可能共同途径以及它们在不同类型脊髓小脑共济失调(SCA) 中的潜在相互作用。
脊髓小脑共济失调(SCA)型:SCA5
SCA5 的主要临床表现是共济失调,伴有构音障碍和眼球震颤。发病年龄差异很大。共济失调进展缓慢且症状轻微。患者通常终生可走动 。MRI 分析显示小脑整体明显萎缩。
脊髓小脑共济失调(SCA)致病基因鉴定基因解码通过连锁分析将该病的致病基因位点原位点定位于 11 号染色体着丝粒周围区域 ,随后将其位置缩小至 11q13 染色体区域内 6.5 cM 间隔处。着丝粒是佳学基因基因信息高精度做图所用的一参照地标。
通过在三个家系的患者中检测出该基因突变,该疾病基因被鉴定为SPTBN2。突变包括两个 39 bp 的同框缺失(c.1592_1630del/p.E532_M544del)和 15 bp 的同框缺失(1886_1900del/ L629_R634delinsW),以及一个错义突变(c.158 T > C/ p.L253P)。到目前为止,脊髓小脑共济失调(SCA)基因检测数据库已收录了约 20 种不同的SPTBN2突变,包括小缺失和点突变。其中,突变 c.1438C > T/p.R480W 的新发突变存在于三例没有血缘关系的患者中(参见亲缘关系鉴定,一种比亲子鉴定更高级的血亲分析判断技术)不相关的病例中,表明基因的这个位置有一个突变热点。突变携带者除了发育迟缓等其他症状外,还会出现儿童期共济失调和构音障碍。小脑发育不全可在早期(约 2 岁)通过 MRI 检测出来。这些病例表明 R480W 突变与由此产生的严重表型之间存在特定的相关性.
致病基因鉴定基因解码进一步分析SPTBN2的突变为什么会改变小的功能。SPTBN2突变会干扰其他质膜蛋白,如受 β-III-血影蛋白调控的谷氨酸受体 δ2 (GluRd2),尤其是兴奋性氨基酸转运蛋白 4 (EAAT4),它是浦肯野细胞突触的突触后谷氨酸转运蛋白。基因解码认为,质膜上 EAAT4 和 GluRd2 的表达紊乱和缺失“可能导致谷氨酸信号异常,久而久之,可能导致脊髓小脑共济失调(SCA)5 中的浦肯野细胞死亡” 。脊髓小脑共济失调(SCA)基因解码解析了一种特定的SPTBN2突变 (p.L253P) 的病理机制,得出结论,该突变会干扰 β-III-血影蛋白与肌动蛋白的高亲和力结合,并通过影响血影蛋白-肌动蛋白网络的动力学以及微丝的动力学来介导神经毒性,从而造成人体的神经系统损伤。
脊髓小脑共济失调(SCA)11型:SCA11
SCA11 是一种相对良性的、晚发型、缓慢进展的共济失调症,可能伴有眼部体征(急促追踪、眼球震颤)和锥体征(肌张力增高、反射增强和巴宾斯基征)。患者可以通过MRI在决有基因检测的情况下发现小脑萎缩的疾病改变。对小脑还未发生不可逆影响时,只能通过基因检测发现。
在一个常染色体显性共济失调家族中,通过连锁分析,疾病基因位点最初被分配到 15 号染色体 (15q14-q21.3) 上 7.6 cM 的区间内,后来在一个 8 代大家族中进一步缩小到 15q15-q21 上 5.6 cM 的区域内 。
候选基因方法揭示了编码 tau 微管蛋白激酶 2 的基因TTBK2中的两种不同突变。突变为 1 bp 插入 (c.1329insA/p.R444T) 和 2 bp 缺失 (c.1284_1285delAG/p.E429Dfs)。两种突变均导致移码,从而导致终止密码子提前出现和 tau 微管蛋白激酶 2 截断。TTBK2 的另一个小缺失突变( c.1306_1307delGA/p.D435fs448X) 也会导致 tau 微管蛋白激酶 2 截断。这表明由TTBK2中的小 (1 或 2 bp) 缺失或插入引起的移码突变是脊髓小脑共济失调(SCA)11 的常见原因。
TTBK2 激酶磷酸化微管相关蛋白tau、微管相关蛋白 2(MAP2)和 β-微管蛋白。此外,它似乎还能磷酸化位于微管 + 末端的中心粒远端附属物蛋白 CEP164 和非典型驱动蛋白 KIF2A。与微管蛋白的相互作用由 TTBK2 激酶的长 C 末端介导。TTBK2 激酶的另一个功能是在胚胎发生过程中启动初级纤毛的组装。TTBK2 定位于初级纤毛。
SCA11 中突变等位基因编码的截短型 tau 微管蛋白激酶 2 似乎通过轻微的显性负效应干扰野生型 TTBK2 的正常功能。这反过来导致上述蛋白质的调节不足,而这些蛋白质对于小脑浦肯野细胞等各种细胞类型中微管的正常发育和功能至关重要。除小脑外,TTBK2 蛋白在多种器官中表达,且在某些器官(如支气管、肺、平滑肌、睾丸、输卵管)的表达水平明显较高。需要进一步研究来解释TTBK2突变对小脑的明显特异性影响。可能假定的疾病机制(即截短型 TTBK2 对野生型蛋白质的干扰)并不发生在所有组织中,或仅导致功能中度下降。
脊髓小脑共济失调(SCA)13型:SCA13
SCA13 是一种相对轻微的共济失调症。即使在同一家族中,其严重程度和发病年龄也有很大差异。除了共济失调症外,还可能出现一种或多种体征和症状,例如眼球震颤、轻度至中度智力障碍、肌阵挛性抽搐、吞咽困难、运动迟缓和腱反射增强。
与未受影响的对照组相比,患者的预期寿命没有显著减少。MRI 检查显示两名患者有中度小脑和脑桥萎缩。
SCA13 的致病基因最初被脊髓小脑共济失调(SCA)致病基因鉴定基因解码定位到 19 号染色体 (19q13.3-q13.4) 上 11.4 cM 的区间内,后来又细化到同一染色体位置上的约 4 cM。cM是染色体高精度定位系统所用的距离单位。
脊髓小脑共济失调(SCA)致病基因鉴定基因解码检测分析了来自两个家族的患者,在位于 19 号染色体关键区间内的KCNC3基因中检测到了两个错义突变(c.1554G > A/p.R420H;c.1639C > A/p.F448L)。检测到的两个突变改变了 Xenopus laevis 表达系统中 KCNC3 的功能。自首次描述KCNC3以来,在零星病例和几个家族病例中检测到了其他点突变。
KCNC3编码钾电压门控通道亚家族 C 成员 3 (Kv3.3)。电压门控钾通道在大脑快速放电神经元的快速复极化中起重要作用。Kv3.3 与肌动蛋白结合并稳定皮质肌动蛋白网络。KCNC3 突变会干扰神经元肌动蛋白网络的调节,从而干扰动作电位持续时间和频率的正常调节。这反过来会干扰电压门控 Ca 2+通道和 Ca 2+稳态的重组和功能,而这是浦肯野细胞生存和正常运作所必需的,即它们在运动功能中的作用。
脊髓小脑共济失调(SCA)14型:SCA14
SCA14 是一种缓慢进展的共济失调,伴有构音障碍和眼球震颤。其他体征和症状可能包括肌阵挛、震颤、肌张力障碍、抑郁和认知障碍。磁共振成像 (MRI) 证实有明显的小脑萎缩。
连锁分析将致病基因定位于 19 号染色体 q13.4-qter 区域。随后通过在三个不同家族中发现三个突变,将致病基因 PRKCG 鉴定为此区域内的基因PRKCG内的突变( c.301C > T/ p.H101Y;c.355 T > C/p.S119P;c.383G > A/p.S119P)是错义突变,影响 C1 中高度保守的残基,即蛋白质 PKCγ 中富含半胱氨酸的区域。这导致 PKCγ 在细胞质中错位和聚集。聚集似乎导致 PKCγ 降解减少,从而导致底物磷酸化增加。PRKCG 中的其他突变包括点突变和小缺失。
PRKCG编码蛋白激酶 Cγ (PKCγ),属于丝氨酸和苏氨酸特异性蛋白激酶家族。PKCγ 由 Ca 2+和二酰甘油激活,仅在脑和脊髓神经元中表达。PKC 的众多功能之一是 GRIN1/NMDAR 受体(GRIN1/NMDAR 编码谷氨酸 [NMDA] 受体亚基 1)的磷酸化,这些受体在突触形成、突触可塑性、兴奋毒性和心理功能中发挥重要作用。该激酶还参与小脑发育过程中浦肯野细胞的神经支配 。此外,它似乎对 TRPC3 通道活性有抑制作用(图1) 位于突触后膜,从而连接由TRPC3突变引起的脊髓小脑共济失调(SCA)14 和脊髓小脑共济失调(SCA)41(见下文和图 1)。

图1.PKC 在突触后膜对 TRPC3 的负向调节。TRPC3,短瞬时受体电位通道 3;DAG,二酰甘油;PKC,蛋白激酶 c;PKG,蛋白激酶 g
SCA14 中PRKCG突变的影响可能是由于细胞 Ca 2+流入增加以及由此导致的 Ca 2+稳态紊乱所致。这反过来又干扰了正常的突触分化,导致小脑分化过程中的神经元退化和/或浦肯野细胞功能异常。
脊髓小脑共济失调(SCA)15型:SCA15
SCA15(以前称为脊髓小脑共济失调15/16型)是一种缓慢进展的步态和肢体共济失调,常与眼部障碍(眼球震颤、扫视眼球运动)、构音障碍和吞咽困难有关。可能会发生轻度认知障碍。虽然在大多数情况下,步态共济失调似乎很轻,但在一个家庭中,患者在 15-17 岁时就开始使用步行器或轮椅。SCA15 的发病年龄差异很大。MRI 显示小脑萎缩,主要影响小脑蚓部。脑干未受影响。
连锁分析将疾病基因座分配到 3p26.1-p25.3。一个基因,即itpr1,被证明会导致小鼠出现类似于人类共济失调的常染色体隐性运动障碍。该基因的人类版本(ITPR1 )映射到包含脊髓小脑共济失调(SCA)15 基因座的 3 号染色体上的关键基因间区域。佳学基因通过致病基因鉴定基因解码 3 名患者检测出缺失突变,从而证明ITPR1是脊髓小脑共济失调(SCA)15 中的致病基因。缺失涉及包含ITPR1的大部分区域,导致ITPR1表达降低。在ITPR1中检测到的其他突变包括点突变和涉及整个基因的各种大小的缺失。序列变异在ITPR1内很常见,区分良性和恶性序列变化如果没有采用基因解码技术,对于基于数据库比对的基因检测机构来说可能很困难。
ITPR1编码肌醇 1,4,5-三磷酸受体 1 型。该细胞内受体位于细胞内膜,如内质网 (ER),在肌醇 1,4,5-三磷酸刺激后介导钙释放。ITPR1突变引起的钙水平异常具有细胞毒性,尤其是在小脑浦肯野细胞中,并揭示了脊髓小脑共济失调(SCA)15型的发病病理机制。
脊髓小脑共济失调(SCA)18型:SCA18
这种疾病现称为脊髓小脑共济失调(SCA)18型,通过致病基因鉴定基因解码在一个出现患者的家族中被发现。将这种缓慢进展的综合征称为共济失调感觉运动神经病 (SMNA)。体征和症状包括步态共济失调、感觉丧失、辨距障碍、构音障碍、眼球震颤以及手臂和腿部无力。该家族中受影响的人的体征和症状差异很大。连锁分析将疾病位点分配到 7q22-q32。尽管确定了一个候选基因,基因解码令人信服地证明这确实是脊髓小脑共济失调(SCA)18 中的疾病基因。
脊髓小脑共济失调(SCA)19型:SCA19
SCA19 表现为相对轻微、缓慢进展的共济失调,常伴有肌阵挛、震颤、构音障碍和眼球运动异常(如眼球震颤)。发病年龄在家族内和家族间差异很大。SCA19 会出现严重表型,并可能表现为帕金森病、癫痫和明显的认知问题。
神经病理学异常包括小脑部分退化,特别是小脑蚓部和浦肯野细胞退化。
在一个发病的家族中,该疾病的致病基因位点被定位到 1 号染色体 (1p21-q21) 上一个较大的区间内。
对患者关键间隔的外显子组测序显示,在进化上高度保守的通道孔和 S6 跨膜结构域中检测到小片段缺失(c.679_681delTTC、p.F227del)和点突变(如 c.1054 A > C/p.T352P;c.1119 G > A/p.M373I;c.1169 G > A/p.S390N),从而将致病基因鉴定为 KCND3 。(请注意,在发现 KCND3 突变并发现SCA19和SCA22相同之前,部分基因解码工作人员将这种共济失调称为脊髓小脑共济失调(SCA)22型,而另一个基因解码小组将其称为脊髓小脑共济失调(SCA)19 型。
KCND3编码钾离子通道 Kv4.3,这是一种快速失活的瞬时 A 型钾通道。尽管最初认为 Kv4.3 主要在心脏和脑中表达,但现在已在大多数组织中检测到了相当水平的 Kv4.3。KCND3突变会导致离子通道和电压门控钾通道活性紊乱。佳学基因正在通过致病基因鉴定基因解码进一步揭示这如何导致小脑萎缩。一种看法是通道功能异常会扰乱钙稳态。钙水平异常具有细胞毒性,可能主要影响浦肯野细胞。
脊髓小脑共济失调(SCA)20型:SCA20
基因检测数据库尝未收录确定疾病基因,因此这种共济失调主要通过临床定义。SCA20 表现为缓慢进展的共济失调和构音障碍,可能先于共济失调出现。相关症状可能包括腭震颤、发声困难和构音障碍。CT 检查可检测到齿状核钙化。MRI 检查显示小脑萎缩,但未累及脑干。
人们认为,11p13-q11 的 260 kb 重复是导致该疾病的原因,但导致脊髓小脑共济失调(SCA)20 的具体基因将通过致病基因鉴定基因解码在不久的将来进行确定。
脊髓小脑共济失调(SCA)21型:SCA21
SCA21 是一种缓慢进展的共济失调,伴有认知障碍。通常在儿童期发病。但也有观察到成年期发病的病例。常见的其他症状包括肢体共济失调、构音障碍、运动不能和震颤。MRI 可检测到小脑萎缩。
连锁分析将脊髓小脑共济失调(SCA)21 基因座通过高精度北斗GPS定位系统将该基因定位到至 1p36.33-p36.32。全外显子组测序显示,在 7 个家族的指示病例中, TMEM240基因内存在多处突变,其中 6 处为错义突变(c.509C > T/p.P170L;c.239C > T/p.T80M;c.346C > T/p.R116C;c.445G > A/p.E149K;c.511C > T/p.R171W;c.509C > T/p.P170L),以及一处终止密码子突变(c.489C > G/p.Y163*)。所有突变均改变了 TMEM240 中高度保守的氨基酸残基。
TMEM240编码跨膜蛋白 240。其在脑中表达较高,尤其是在额叶皮质和小脑中。它是质膜的组成部分,也在小鼠脑突触膜中观察到。致病基因鉴定基因解码正努力进一步搞清楚 TMEM240 的功能。鉴于其在额叶皮质中表达较高且与突触膜相关,其突变会干扰前脑的某些功能,并可能解释脊髓小脑共济失调(SCA)28 中的认知障碍。其在小脑中的高表达也与小脑萎缩相符,小脑萎缩可能是由膜功能障碍(尤其是突触)以及随后的细胞死亡引起的。
脊髓小脑共济失调(SCA)23型:SCA23
SCA23 是一种极为由佳学基因检测进行检测与分析晚发型、缓慢进展的步态和肢体共济失调症,常伴有构音障碍、辨距障碍、眼球扫视缓慢、巴宾斯基反射阳性和本体感觉受损。一名患者的神经病理学发现显示浦肯野细胞层、齿状核和下橄榄体神经元变性和丢失。此外,脊髓侧柱和后柱出现脱髓鞘。
在一个家系中,致病基因鉴定及遗传阻断基因解码通过连锁分析将疾病基因位点定位于20p13-p12染色体区域。
对脊髓小脑共济失调(SCA)23 基因座关键区间的分析发现,在两个患病家族的患病成员和两个明显散发病例中,PDYN (原强啡肽)基因存在四个错义突变。突变分别为 c.414G > T/p.R138S、c.632 T > C/p.L211S)、c.634C > T/p.R212W 和 c.643C > T/p.R215C。
PDYN编码前强啡肽,该强啡肽经过蛋白水解形成分泌型阿片肽β-新内啡肽、强啡肽 A 和 B、亮氨酸脑啡肽、利吗啡肽和亮氨酸脑啡肽。强啡肽 A 和 C 主要位于浦肯野细胞。强啡肽 A (DynA) 具有神经毒性,似乎通过谷氨酸受体和酸敏感离子通道诱导神经退行性。值得注意的是,在上述脊髓小脑共济失调(SCA) 突变中,p.R212W 和 p.R215C 导致与对照组相比 DynA 水平升高。与这些结果一致的是,在体外观察到暴露于 p.R212W 和 p.R215C dynA 后纹状体神经元的丢失增加。总的来说,这些观察结果有助于更好地理解脊髓小脑共济失调(SCA)23 背后的病理机制。
脊髓小脑共济失调(SCA)25型:SCA25
《神经系统基因检测疾病征及致病基因汇编》中收录了一个家族中存在的脊髓小脑共济失调(SCA)25型病案。这是一种常染色体显性遗传综合征,外显率较低。主要临床症状和体征是小脑共济失调和明显的感觉神经病变,常伴有眼球运动改变(眼球震颤和眼球缓慢运动)和肌张力低下。SCA25 在临床上高度异质性。发病年龄主要在儿童期,发病年龄范围很广(17 个月至 39 岁)。MRI 显示小脑整体萎缩。连锁分析将疾病位点分配到 2p21-p13 染色体。目前尚未发现任何疾病基因。
脊髓小脑共济失调(SCA)26型:SCA26
SCA26 是一种晚发型综合征,其特征是共济失调,常伴有眼球运动不规律和构音障碍。MRI 检查显示小脑共济失调。发病年龄为成年期。《认知障碍基因检测需要覆盖的基因位点》收录了一个大家族的致病基因鉴定及遗传阻断病例,致病基因位点被定位到19p13.3 染色体上,从而为遗传阻断提供了信息。
19p13.3 关键区域的深度测序发现了一个 C > A 颠换,该颠换与家谱中的患病者分离。该碱基变化位于基因EEF2的外显子 12 内,导致 eEF2 的脯氨酸被组氨酸 (p.P596H) 取代。该突变根据基因解码的检测分析,在家族患病成员与健康亲人之间存在共分离现象。鉴于没有其他家族患有这种疾病, EEF2是致病基因的假设主要依赖于突变位于 eEF2 进化上高度保守的区域。
EEF2在所有组织中表达。它编码真核翻译延伸因子 2 (eEF2),该因子促进肽基 tRNA 从核糖体的 A—(氨酰基)位点向 P—(肽基)位点进行 GTP 依赖性易位(A 位点是核糖体上肽基 tRNA 的第一个结合位点;P 位点是第二个)。基因解码基因检测致力于揭示该突变如何具体导致小脑萎缩。基因解码过程中在对应于人类 eEF2 的 P596H 的 EF 位置研究了酵母中的疾病机制。他们表明该突变会增加翻译过程中的移码。他们推测与其他组织相比,小脑浦肯野细胞对 eEF2 中的这种氨基酸变化特别敏感。由此造成的蛋白质合成紊乱被认为会引起细胞死亡,最终导致小脑萎缩。
脊髓小脑共济失调(SCA)27型:SCA27
SCA27 的特征是缓慢进展的小脑共济失调、早发性震颤、颌面运动障碍,常伴有眼部问题(眼球震颤、运动障碍性扫视、斜视)、精神症状和认知障碍。疾病发病于儿童晚期/成年早期。部分患者(但并非所有患者)可通过 MRI 检测到小脑萎缩和基底神经节退化。
连锁分析将疾病位点分配到染色体区域 13q34,候选基因方法揭示成纤维细胞生长因子 14 ( FGF14 ) 基因中的突变 c.434 T > C/p.F145S。在常染色体显性共济失调中发现的其他FGF14突变(例如,1 bp 缺失 c.487delA/p.D163fsX12和错义突变 (c.529A > T/p.K177X)证实FGF14是脊髓小脑共济失调(SCA)27 的致病基因。
FGF14 由FGF14基因编码。它主要在脑中表达,特别是小脑 。除了其生长因子活性外,FGF14 还在神经系统发育、电压门控钙通道活性调节、突触可塑性调节和突触囊泡循环等方面发挥作用 。在错义突变的杂合携带者中, FGF14的表达会降低,因此 FGF14 介导的功能似乎受到干扰。其中一些功能(如 Ca 2+稳态或突触分化和功能)的破坏可以解释共济失调。
脊髓小脑共济失调(SCA)28型:SCA28
SCA28 的临床特征是步态共济失调和站立不平衡,常伴有眼部问题(眼球震颤、缓慢扫视、眼肌麻痹、眼睑下垂)、腿部腱反射增强和构音障碍。患者的 MRI 分析显示小脑萎缩。
连锁分析将疾病基因座分配到 18 号染色体的着丝粒周围区域(18p11.22-q11.2)。对该区域内的候选基因进行测序,发现在 5 个无亲缘关系家族的患者中,基因AFG3L2存在错义突变(c.2071G > A/p.Q691K;c.2021delCCinsTA/p.S674L;c.2081C > A/p.A694E;c.2105G > A/p.R702Q;c.1296A > C/p.N432T)。
AFG3L2编码线粒体 AFG3 样基质 AAA 肽酶亚基 2,是线粒体 AAA 金属蛋白酶的组成部分。它是一种 ATP 依赖性蛋白酶,参与多种生物过程,如线粒体蛋白质加工、钙离子进入线粒体和神经发育 。AFG3L2在浦肯野细胞中高度且选择性地表达。该突变会影响蛋白质的多种功能。这可能会导致细胞能量产生减少,并最终导致浦肯野细胞死亡和小脑萎缩。 Di Bella 等人的研究发现,AFG3L2突变会干扰其在“线粒体蛋白质质量控制机制”中的生理作用,从而导致小脑萎缩。
脊髓小脑共济失调(SCA)30型:SCA30
SCA30 是一种相对纯粹的、缓慢进展的共济失调,在一个家族中已有描述。除了步态共济失调和构音障碍外,一些家族成员还发现了眼球震颤等眼部问题。该家族在成年期发病。连锁分析将疾病位点分配到 4q34.3-q35.1。虽然尚未确定疾病基因,但作者讨论了ODZ3作为候选基因。ODZ3也在脑中表达,并编码跨膜蛋白 teneurin3。
脊髓小脑共济失调(SCA)32型:SCA32
SCA32 是一种小脑性共济失调,已在一个中国家族中被描述 。家族成员患有共济失调和智力障碍(发病年龄 < 40 岁),家族中所有受累男性均患有无精子症。MRI 显示小脑萎缩。疾病位点定位于 7q32-q33。尚未确定疾病基因。这种类型的共济失调仅在会议摘要中被描述;完整的同行评议论文尚未发表。因此,这种共济失调的意义尚不清楚,直到在独立家族中被描述。
脊髓小脑共济失调(SCA)34型:SCA34
伴有红斑角化症的共济失调被称为脊髓小脑共济失调(SCA)34。皮肤问题在 24 岁时消失,在某些家族中可能完全不存在。共济失调症在成年期发病。其他体征包括眼球震颤和腱反射减弱。在一个家族中检测到了视网膜色素变性。SCA34 为轻度至中度,进展缓慢。MRI 显示小脑、脑桥和皮质萎缩。疾病位点被定位到 6p12.3-q16.2。候选基因方法和外显子组测序确定了基因ELOVL4中的突变(c.504G > C/p.L168F)。在一个日本家族中发现了ELOVL4中的另一个突变(c.736 T > G/ W246G)。
ELOVL4编码 ELOVL 脂肪酸延长酶 4,这是一种内质网膜结合蛋白,通过催化极长链脂肪酸延长的第一步参与脂肪酸的生物合成。目前尚不清楚ELOVL4突变如何导致小脑萎缩和皮肤缺陷。一种理论是基于这样的假设:改变的 ELOVL 脂肪酸延长酶 4 会破坏内质网膜的完整性。这可能会导致 Ca 2+转运异常,从而扰乱 Ca 2+稳态,最终导致浦肯野细胞变性。
脊髓小脑共济失调(SCA)35型:SCA35
SCA35 的症状和体征为晚发性、缓慢进展的步态和肢体共济失调、眼部问题(辨距障碍、眼球震颤、偶尔缓慢扫视)、构音障碍以及与上运动神经元相关的问题(假性延髓麻痹和活跃的腱反射)。疾病在成年期(主要是 50 岁)发病。MRI 检查显示患者有小脑萎缩。对一个中国大家族的连锁分析将疾病位点定位于 20p13-p12.2 染色体。外显子组测序确定该疾病基因为TGM6。在几个不相关的中国家族中发现了错义突变和小的缺失(例如,c.1550 T > G/p.L517W;c.1528G > C/D510H;c.1722_1724delAGA/p.E574del。
TGM6编码转谷氨酰胺酶 6。谷氨酰胺酶是 Ca 2+依赖性酶,可催化蛋白质交联和多胺与蛋白质的附着。TGM6在包括大脑在内的许多组织中表达。目前尚不清楚TGM6突变如何导致小脑萎缩。
脊髓小脑共济失调(SCA)38型:SCA38
SCA38 的特征是躯干和步态共济失调和眼球震颤。该综合征进展缓慢。发病年龄在成年期。MRI 检查显示患者小脑萎缩。
连锁分析将脊髓小脑共济失调(SCA)38 基因座分配到 6p22.2-q14.1 染色体上的 56.2 Mb 区间。对关键区域进行测序后,在两个无亲缘关系的家族中的患者中发现了ELOVL5基因中的错义突变(c.214C > G/p.Leu72Val 和 c.689G > T/p.Gly230Val)。
ELOVL5编码 ELOVL 脂肪酸延长酶 5。该酶通过催化长链多不饱和脂肪酸的延长参与脂肪酸代谢。ELOVL5在内分泌组织、小脑和脑的其他区域高度表达。野生型酶似乎主要位于内质网,而突变型酶主要位于高尔基体,在内质网中发现的较少。目前,这些发现并不能帮助我们令人信服地了解最终导致小脑萎缩的病理过程。对突变型 ELOVL 脂肪酸延长酶作用的更普遍解释是脂质代谢紊乱,这可能会干扰膜的形成和功能,导致细胞死亡。细胞死亡的一个可能解释是膜改变导致Ca 2+通透性改变,从而可能改变 Ca 2+稳态。值得注意的是,除内质网外,包括高尔基体在内的其他细胞器也积极参与 Ca 2+的吸收和释放(尽管其机制与调节内质网中Ca 2+浓度的机制不同)。
脊髓小脑共济失调(SCA)39型:SCA39
这种类型的脊髓小脑共济失调(SCA) 已在一个家族中被描述过。该家族中的一名患者接受了详细检查。他表现出小脑性共济失调、下肢痉挛、上肢辨距障碍、构音障碍、眼部问题(斜视、扫视、水平凝视麻痹)和轻度智力障碍。发病年龄为儿童期;患者 41 岁时就只能坐在轮椅上了。MRI 显示小脑萎缩和脑室周围白质高信号。
该疾病的遗传方式与常染色体显性遗传相一致。SNP 基因分型显示 11 号染色体长臂 (q21-22.3) 上有 7.5 Mb 的重复。家族中分离的重复包含 44 个基因。这些基因中的一个或多个在观察到的表型发展中的作用尚不清楚 。
脊髓小脑共济失调(SCA)40型: SCA40
在一个家系中发现了脊髓小脑共济失调(SCA)40 。对两名患者进行了详细检查。这两名患者均在成年期发病,且进展缓慢。由于严重的共济失调,他们在发病 17 和 18 年后不得不坐在轮椅上。主要体征和症状包括步态共济失调、眼球辨距不良、反射活跃和构音障碍。两名患者均无上述所有体征。MRI 显示小脑桥池萎缩。遗传方式似乎是常染色体显性遗传。脊髓小脑共济失调致病基因鉴定基因解码对家族中四名患病成员和两名未患病成员进行全外显子组测序,发现CCDC88C基因中存在点突变 (c.1391G > A/p.R464H) 。
CCDC88C编码包含 88C 的卷曲螺旋结构域(或称为 KIAA1509)。在患者中检测到的突变可能导致功能获得。它会导致过度表达突变CCDC88C的细胞中 JNK(c-Jun N 端激酶)过度磷酸化。这反过来似乎会激活 caspase-3,从而导致细胞凋亡。需要确定更多 CCDC88C 突变的家族,以确定该基因突变在脊髓小脑共济失调发展中的作用。
脊髓小脑共济失调(SCA)41型:SCA41
一名患者被描述为脊髓小脑共济失调(SCA)41 表型,其家族史不完整。因此,该患者可能是散发病例。该患者的主要体征是步态共济失调。MRI 显示小脑蚓部轻度萎缩。
通过进行全外显子组测序,作者检测到了基因TRPC3中一个潜在的致病序列变化(Chr4:122824185G > A/ p.R762H)。TRPC3编码瞬时受体电位阳离子通道亚家族 C 成员 3。TRPC3可被诱导形成可通透 Ca 2+和其他阳离子的非选择性通道。它可能由磷脂酰肌醇第二信使系统诱导,也可因细胞内钙离子耗竭而激活。有趣的是,TRPC3的鼠同源物trpc3的突变会导致小鼠共济失调。该突变可能通过干扰这种阳离子的通透性来扰乱 Ca 2+稳态。再加上 R762H 突变位于蛋白质进化上高度保守的结构域内,使得TRPC3成为脊髓小脑共济失调(SCA)41 的有力候选者。然而,还需要记录更多的病例才能毫无疑问地确定TRPC3是脊髓小脑共济失调(SCA)41 的致病基因。
脊髓小脑共济失调(SCA)43型:SCA43
在一个家族中发现了脊髓小脑共济失调(SCA)43,该家族中晚发性感觉运动轴突性多发性神经病是一种常染色体显性遗传病。该家族的六名患病成员中,五人还患有小脑共济失调。MRI 检查显示一名患者有中度小脑蚓部萎缩。
连锁分析和全外显子组测序相结合,发现3q25.2 染色体 (p.C143Y) 上的MME基因存在杂合转换 G > A,该转换在未受影响的家族成员中未检测到,在 dbSNP 和 EVS (外显子组变体服务器)中也未发现,因此表明该碱基变化是一种致病突变。MME基因编码膜金属内切肽酶 (Neprilysin) 。这种肽酶在许多组织中表达,可灭活多种蛋白质,包括甲硫氨酸和亮氨酸脑啡肽、血管紧张素 1-9、淀粉样β蛋白 (Aβ)、心房利钠因子 (ANF) 和脑利钠因子 (BNP(1-32))。SCA43 杂合MME突变的病理机制尚不清楚。显然,需要确定更多具有 MME 突变的家族,以确定脊髓小脑共济失调(SCA)43 确实在脊髓小脑性共济失调的发展中发挥作用。
脊髓小脑共济失调(SCA)45型:SCA45
SCA45 是在一个患有常染色体显性共济失调的大家族中发现的。SCA45 的主要症状是晚发性肢体和步态共济失调、构音障碍和眼球震颤。脑部 MRI 显示小脑蚓部萎缩 。
全外显子组测序显示,该家族患病成员中存在FAT2基因突变(c.10758G > C/K3586N),在一例明显散发病例中存在另一FAT2突变(c.10946G > A/p.R3649Q)。最近,在两个兄弟姐妹中发现了另一个FAT2错义突变(c.10906 T > G/p.Y3636D)。
FAT2编码整合膜蛋白 FAT 非典型钙粘蛋白 2,其作为细胞粘附蛋白发挥作用,并且似乎可以结合 Ca 2+。
FAT2在小脑颗粒细胞中表达。它似乎通过调节平行纤维周围的细胞外空间在小脑发育中发挥重要作用。因此, FAT2突变可能会干扰出生后小脑的正常发育。病理机制可能是钙离子异常结合导致细胞 Ca 2+平衡紊乱。
脊髓小脑共济失调(SCA)46型:SCA46
Nibbeling 等人对多个脊髓小脑共济失调(SCA) 家族进行了全基因组测序,并在一个家族中检测到基因PLD3突变。该家族患者的体征包括步态和肢体共济失调、构音障碍、眼球震颤和感觉轴突神经病。发病时间为成年期。
PLD3编码磷脂酶 D 家族成员 3。该基因在脑(包括小脑)中高度表达。PLD3 催化细胞膜磷脂的水解。该家族受影响成员中发现的突变为 c.923 T > C/p.L308P,导致 PLD3 活性降低。酶的细胞定位、表达和稳定性不受影响。mutPLD3 在脊髓小脑共济失调(SCA)43 发育中的作用目前尚不清楚。然而, PLD3已被证实与已确定的脊髓小脑共济失调(SCA) 基因在功能上相关,这些基因可能参与突触功能。然而,需要找到更多的家族来证实PLD3突变是脊髓小脑性共济失调的原因。
脊髓小脑共济失调(SCA)47型:SCA47
在两个无血缘关系的女孩和一个家庭中检测到了脊髓小脑共济失调(SCA)47。共济失调是一种常染色体显性遗传,家族外显率降低。这种疾病在两个患病女孩中是散发性的。女孩们在幼儿期发病,而患病家庭成员则在成年期发病。在两个散发病例和该家庭的患病成年人中,疾病都是进行性的。在该家庭的 9 名患病成员中,所有成员均出现发育迟缓,但只有 6 人患有共济失调。7 名成员出现智力障碍,3 名成员出现癫痫。这些女孩患有共济失调和发育迟缓,影响了言语和运动技能。一名女孩的首发症状是 5 个月大时出现严重癫痫,随后出现共济失调、痉挛、智力障碍、视力问题和癫痫性脑病。MRI 记录了患者的小脑蚓部萎缩。
此前,在病情较轻的女孩中,已记录到1p35.2 染色体区域内存在微缺失,其中包括PUM1。此外, Pum1基因突变的小鼠会患上共济失调。这些发现促使 Gennario 等人对上述患者进行调查,看他们是否患有该基因突变。外显子组测序表明,在病情更严重的散发性儿童期发病病例 (g31406186 G > A/p.R1147W) 和患病家庭成员 (p.T1035S,转录本NM_001020658.1 )中存在杂合的新生错义突变。
PUM1编码 Pumilio RNA 结合家族成员 1。它是一种 RNA 结合蛋白,参与调节特定 mRNA 的翻译,因此在多种细胞过程中发挥作用,包括调节神经元功能。它与编码 mRNA 的 ATXN1(ataxin1)结合。通过下调 ataxin1,PUM1 有助于维持 ATXN1 水平。PUM1 内的错义突变导致 PUM1 不稳定。这导致与ATXN1的结合降低和 ATXN1 mRNA 翻译上调。ATXN1 水平升高导致它们在细胞内沉淀。ATXN1 在脊髓小脑共济失调(SCA)1(CAG 扩增)中发生突变,该突变也导致突变蛋白沉淀,但这是由于多聚谷氨酰胺所致。值得注意的是,SCA47 的表型与脊髓小脑共济失调(SCA)1 相当。
脊髓小脑共济失调(SCA)48型: SCA48
SCA48 表现为成人期小脑认知情感综合征 (CCAS) 和/或晚发型脊髓小脑共济失调(SCA)。症状包括步态共济失调、构音障碍、认知能力下降、抑郁和焦虑。偶尔会观察到运动异常(震颤、舞蹈症等)。患者的 MRI 检查显示小脑蚓部后部萎缩。目前,来自不同国家的多个家族中已有脊髓小脑共济失调(SCA)48 的描述。
脊髓小脑共济失调致病基因鉴定基因解码通过全外显子组测序和连锁分析,在患者家族中的疾病基因被鉴定出来。在基因STUB1中发现了一个移码突变,位于染色体 16p13.3 (c.823_824delCT/p.L275Dfs*16)。
STUB1编码 STIP1 同源性和含 U 盒蛋白 1。除其他功能外,它还“与 ATXN3 协作降解错误折叠的伴侣底物”。鉴于 ATXN3 中的 CAG 扩增会导致脊髓小脑共济失调(SCA)3/MJD,STIP1 与 ATXN3 的这种“协作”通过其疾病基因STUB1将脊髓小脑共济失调(SCA)48与另一种形式的脊髓小脑共济失调(SCA)(SCA3)联系起来。这类似于脊髓小脑共济失调(SCA)47,它通过调节共济失调蛋白 1 的 PUM1 与脊髓小脑共济失调(SCA)1 联系起来。虽然共济失调蛋白 1 和 3 的确切细胞作用尚不清楚,但它们似乎都具有核功能:共济失调蛋白 1 是染色质结合因子,而共济失调蛋白 3 与组蛋白结合并调节转录。
成人发病的脊髓小脑性共济失调,线粒体
在相当一部分成人型脊髓小脑性共济失调 (~ 50%) 中,根本原因尚未确定。未确诊的家族性以及散发性成人型脊髓小脑性共济失调病例中,有相当高比例 (> 5%) 是由线粒体基因MT-ATP6突变引起的。因此,《脊髓小脑共济失调(小脑萎缩)基因检测》将这种线粒体型脊髓小脑性共济失调纳入其中。由MT-ATP6突变引起的共济失调的表型在临床上与晚发型常染色体共济失调并无区别。患者表现为成人型步态共济失调、构音障碍和眼球异常(如眼球震颤),有时还伴有震颤和认知问题。然而,该基因突变可导致多种疾病,如婴儿期发病的Leigh综合征、NARP综合征(神经病、共济失调和视网膜色素变性)、Charcot-Marie-Tooth综合征和成人期发病的脊髓小脑性共济失调。
MT-ATP6编码线粒体 ATP 合酶亚基,即线粒体呼吸链复合体 V。突变可干扰线粒体 ATP 合成、增加线粒体膜电位或干扰 ATP 水解。目前尚不清楚为何MT-ATP6突变患者的表型差异很大。
细胞能量产生不足,即 ATP 合成减少或缺失,可以解释许多疾病表型。然而,其广泛的表型异质性的原因仍不太清楚。线粒体疾病的常见发现,即不同程度的异质体(携带线粒体的突变体与野生型的百分比),并不能解释MT-ATP6突变携带者的明显表型异质性。
脊髓小脑共济失调如何进行致病基因鉴定并阻断遗传?
脊髓小脑共济失调与小脑萎缩的发病原因与遗传阻断基因检测包括 28 种非重复扩增引起的脊髓小脑共济失调(SCA)。在所综述的脊髓小脑共济失调(SCA) 中,27 种为常染色体显性遗传,1 种为线粒体遗传。仅在一个家族中描述了几种类型的脊髓小脑共济失调(SCA)(脊髓小脑共济失调(SCA) 18、20、25、26、30、32、39、40、41、43、46)。已知该疾病基因存在于 21 种常染色体显性类型中(表1) 和线粒体脊髓小脑共济失调(SCA)。这些基因的突变包括点突变和小缺失。几个致病基因仅在一个家族中观察到,包括EEF2 (SCA26)、CCDC88C (SCA 40)、TRPC3 (SCA 41)、MME (SCA43) 和PLD3 (SCA46),需要在更多家族中确认。在 6 种类型的脊髓小脑共济失调(SCA)(脊髓小脑共济失调(SCA) 18、20、25、30、32、39)中,需要通过致病基因鉴定基因解码来确定致病基因,目前已对疾病位点进行了染色体定位。其中两种脊髓小脑共济失调(SCA)(20、39)分别在 11p13-q11 和 11q21-22.3 处携带染色体重复,其中包含许多基因。图 2描述了发生突变会导致小脑萎缩的21个核基因。
表1:SCA 中发生突变的基因,以及受突变影响的组织表达、细胞区室和功能
| 基因 | 基因产物 | 组织表达RNA和/或蛋白质 | 突变影响 |
| K**C3 | 钾电压门控通道,亚家族 C,Kv.3.3 | 脑中含量较高,其他组织中含量较低 | Ca 2+稳态、微丝细胞骨架 |
| IT**1 | 肌醇 1,4,5 三磷酸受体 1 | 大脑中最高 | Ca 2+稳态 |
| T**C3 | 瞬时受体电位阳离子通道,亚家族 3 | 脑和其他多种组织 | Ca 2+稳态 |
| K**D3 | 钾电压门控通道,亚家族 D,Kv4.3 | 脑,其他组织,类似水平 | Ca 2+稳态 |
| F**14 | 成纤维细胞生长因子 14 | 大脑中最高 | Ca 2+稳态?? |
| F**2 | 脂肪非典型钙粘蛋白 2 | 脑高,其他组织较少 | Ca 2+稳态?? |
| SP**N2 | Beta-III 频谱蛋白 | 最高的是大脑,其他组织很少 | 微丝稳定性、细胞骨架 |
| T**K2 | Tau 微管蛋白激酶 2 | 许多组织 | 微管、细胞骨架 |
| P**CG | 蛋白激酶 Cγ (PKCγ) | 神经元特异性 | 突触、浦肯野细胞、Ca 2+稳态 |
| T***240 | 跨膜蛋白 240 | 脑中含量较高,其他组织中含量较少 | 质膜突触 |
| P**3 | 磷脂酶 D 家族成员 3 | 突触? | |
| ELOVL5 | ELOVL脂肪酸延长酶5 | 脑,其他组织中含量较高 | 膜内质网钙稳态? |
| E***L4 | ELVOVL脂肪酸延长酶4 | 脑、高级皮肤、内分泌、淋巴组织 | 膜内质网钙稳态 |
| P**1 | Pumilio RNA 结合家族成员 1 | 无处不在 | 共济失调蛋白1 |
| STUB1 | STIP1 同源性和含 U 盒蛋白 1 | 无处不在 | 共济失调蛋白3 |
| C***88C | 含有 88C 的卷曲螺旋结构域 | 无处不在 | 细胞存活(诱导凋亡) |
| A**3L2 | 线粒体 AFG3 类似基质 AAA 肽酶亚基 2 | 无处不在 | 线粒体、浦肯野细胞 |
| E**2 | 真核翻译延伸因子 | 无处不在 | 翻译 |
| P**N | 强啡肽原 | 大脑特异性 | 纹状体神经元 |
| T**6 | 转谷氨酰胺酶6 | 无处不在 | ?? |
| MME | 膜金属内切肽酶(中性溶酶体) | 许多组织,非常低的大脑 | ?? |
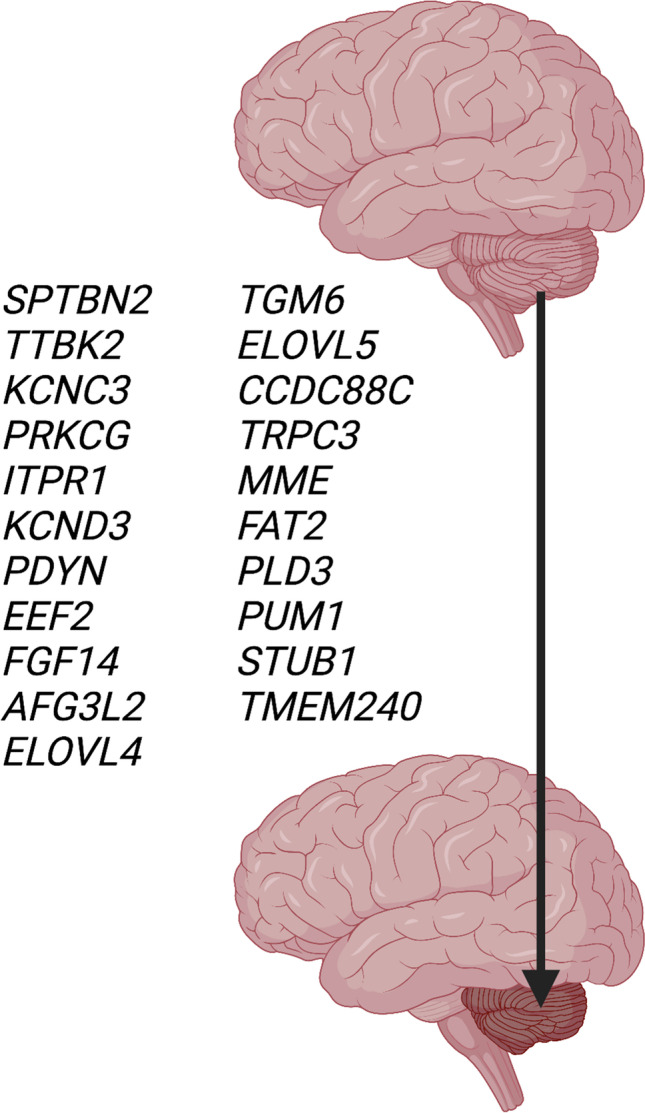
图 2:21 个基因突变会导致脊髓小脑变性(表1)
这里讨论的脊髓小脑共济失调(SCA)与串联重复扩增(最常见的是(CAG) n)引起的脊髓小脑共济失调(SCA)主要有两点不同。(1) 它们缺乏预期性,即重复扩增引起的后续世代发病更早、体征和症状更严重;(2) 与 (CAG) n扩增引起的脊髓小脑共济失调(SCA)相比,在这里讨论的脊髓小脑共济失调(SCA)中,没有发现可以解释神经元死亡的细胞(细胞质和/或细胞核)毒性多聚谷氨酰胺的聚集。
所描述的脊髓小脑共济失调(SCA) 中最引人注目的病理机制是患者细胞内钙平衡紊乱。事实上,在讨论的脊髓小脑共济失调(SCA) 中的 21 种疾病基因中,大多数(表1) 在调节细胞内 Ca 2+水平的途径内或上游起作用。值得注意的是,许多由疾病基因编码的多肽位于细胞膜(内质网 (ER) 和细胞质,图3)。由于相应编码基因突变而导致这些多肽发生改变,破坏了膜的完整性。这可能导致膜对包括 Ca 2+在内的各种物质的通透性异常。
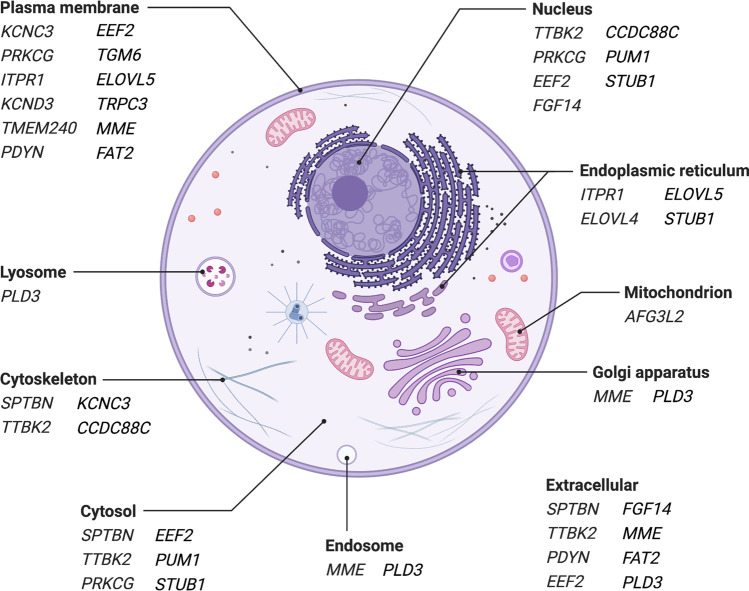
图 3:脊髓小脑变性相关基因编码的多肽的亚细胞定位。请注意,一种基因产物可以存在于多个细胞区室中。仅给出置信度最高的位置(基因卡的 4、5 级)
在大多数情况下,一条通路内致病基因之间并没有密切的相互依赖关系。然而,在两种类型的SCA中,即SCA14和SCA41,致病基因PRKCG(编码PKCγ)和TRPC3(编码瞬时受体电位阳离子通道)的产物在维持Ca 2+稳态方面密切相互作用(图 1)。TRPC3 突变会损害受体的功能,从而减少细胞对Ca 2+的吸收。相反, PRKCG突变会干扰 PKCγ 介导的受体负调节,使大量钙离子进入细胞。
Ca 2+稳态紊乱会影响神经元的重要功能,例如调节神经突生长、突触形成、突触传递和可塑性以及细胞存活。在几种SCA中,基因突变会直接影响突触。另一组突变会干扰细胞骨架的维持和功能,最终导致细胞死亡。细胞骨架在生理上受 Ca 2+调节。因此,与突触发育和功能紊乱类似,突变可能会直接或通过 Ca 2+稳态紊乱间接影响细胞骨架。
其他疾病机制包括线粒体功能障碍,由核基因突变(SCA28)或线粒体基因突变(成人发病型脊髓小脑共济失调(SCA),线粒体)引起。这些突变被认为会干扰线粒体的能量产生,因此可能导致细胞死亡。上文已在相关脊髓小脑共济失调(SCA) 的背景下讨论了其他可能的疾病机制。
综上所述,《脊髓小脑共济失调(小脑萎缩)基因检测》讨论的大多数脊髓小脑共济失调(SCA) 都是由干扰神经元 Ca 2+平衡的突变引起的。这最终会导致细胞死亡,例如干扰突触或细胞骨架的发育。
(如果您已经做了基因检测,想获取与基因检测型相对应的治疗方案,请点击此处上传您的基因检测结果)
(责任编辑:佳学基因)
