












【基因检测标准】眼科遗传病基因诊断方法专家共识
中国眼遗传病诊疗小组 中国眼科遗传联盟 通信作者:睢瑞芳,Email:hrfsui@163.com
DOI:10.3760/cma.j.issn.2095 0160.2018.07.001 基金项目:中国医学科学院医学与健康科技创新工程经费资助项目
(2016-I2M-1-002);国家重点研发计划(2016YFC0901500)
内容导航
1、NGS技术及其应用2、眼遗传病基因检测流程
3、常见眼遗传病及其临床和基因特征
1、NGS技术及其应用
眼遗传病是一组由于基因缺陷导致的眼部疾病。2018年4月27日在人类孟德尔遗传在线数据库(Online Mendelian Inheritance in Man,OMIM)(http://www.omim.org/)中以“eye”作为关键词搜索疾病表型可得到440种疾病条目。临床常见的眼遗传病有视网膜变性、先天性青光眼、先天性白内障、遗传性视神经病变、先天性眼外肌异常及累及眼部的一些综合征等。遗传方式包括常染色体显性遗传、常染色体隐性遗传、X性连锁遗传、双基因遗传及线粒体遗传等。此外,眼遗传病同样存在着等位基因异质性和基因座异质性。同时,不同地域、不同民族之间的基因变异和表型均存在着较大的差异,这些因素为眼遗传病的临床诊断和分子检测带来了巨大的挑战。随着人类基因组参考序列的完成、基因芯片和高通量测序等技术的问世以及生物信息技术对海量生物数据高效分析和处理技术的发展,近几年来单基因遗传病的分子诊断效率迅速提升,技术方法取得了很大的突破,为患者和临床医师的遗传咨询提供了技术保障,更为将来的基因治疗奠定了基础。目前,在众多基因组技术中二代测序(next-generation sequencing,NGS)技术尤以其特有的优势在包括眼遗传病在内的单基因遗传病的研究中发挥着重要作用,并越来越多地用于眼遗传病的分子检测。然而,由于基因检测技术的应用和方法选择在技术层面有一定的难度,更由于眼遗传性疾病有较大的基因突变异质性和临床表型异质性,在临床实践中我们发现对NGS的应用存在偏差,给遗传性眼病诊断结果和患者成本-效益带来一定的问题。为规范基因检测在眼遗传病分子诊断中的应用,我们组织了有关专家根据目前我国的实际情况,在深入分析及了解各种遗传性眼病表型的复杂性和致病基因变异复杂性的基础上进行反复讨论,提出眼遗传性疾病基因分子诊断规范化推荐意见,以供眼科相关临床人员和实验室检测人员在实践中参照应用。
本共识专家组成员由国内外人类眼遗传病专家和全国眼遗传病研究专家组成。共识的制定基于人类遗传性疾病基因分子诊断技术方法的科学性和适用范围,总结相关的推荐意见和观点,注重眼遗传性疾病基因分子诊断的临床实践可操作性和指导性,重点回答以下重要问题:(1) NGS的技术特点、适用范围及其优势和潜在局限性。(2)眼遗传性疾病如何遵循基因检测流程。(3)如何根据眼遗传疾病的临床特征合理选择基因检测方法。(4) 眼遗传病基因分子检测的目的及意义。
1、NGS技术及其应用
使用NGS技术可同时进行大量基因序列的平行测序。NGS分为靶向基因测序(target gene sequencing,TGS)(panel">或称panel测序)、全基因外显子组测序(whole exome sequencing,WES)和全基因组测序(whole genome sequencing,WGS)。Panel测序和WES的基本流程为目标区域基因片段的获得和富集、对捕获片段的扩增和高通量测序、生物信息学分析及验证,贼后确定致病突变。WGS是对全基因组的序列进行测序,故没有捕获这一步骤,可直接将基因组片段打断后进行高通量测序。这3种方法原理相似,但又有各自的特点。此外成本还存在差异,因而在应用范围上也各有优势。目前panel测序和WES广泛用于单基因遗传病的分子检测。
NGS技术对检测人员、实验室标准、试剂及项目选择、实验室质量管理等均有严格的要求,具体可参照《中华病理学杂志》在2017年3月发表的“临床分子病理实验室二代基因测序检测专家共识”。临床基因检测报告作为连接受检者、实验室技术人员和临床医师的重要依据,其内容在国内有也有了行业共识,具体可参考2018年2月《中华医学遗传学杂志》刊出的“临床基因检测报告规范与基因检测行业共识探讨”。该文对遗传病基因检测报告的原则、规范化和标准化提出了建议。结合基本规范,针对眼遗传性疾病已知的相关致病基因采用相应的优化检测模块,选择对外显子及已知内涵子突变更多覆盖的检测方法是提高检测结果高效性的关键。
每种技术的临床应用过程中都存在不足,NGS也存在着一些缺点:(1)测序错误率高于传统的一代测序技术,需要增加测序深度、提高覆盖率等方法来补偿。(2)由于技术局限,对基因组鸟嘌呤和胞嘧啶所占比率高(GC含量)的目标区域捕获率达不到100%。(3)如果测序片段较短,高重复区域检测正确度下降。(4)对拷贝数变异(copy number variants,CNVs)、动态突变、复杂结构重排等变异类型的检测存在局限性。总之,在了解NGS优缺点的基础上,使其在临床单基因遗传病的分子检测中得到更合理的应用是目前基因检测过程中检测人员和临床医师面临的挑战[1]。
2、眼遗传病基因检测流程
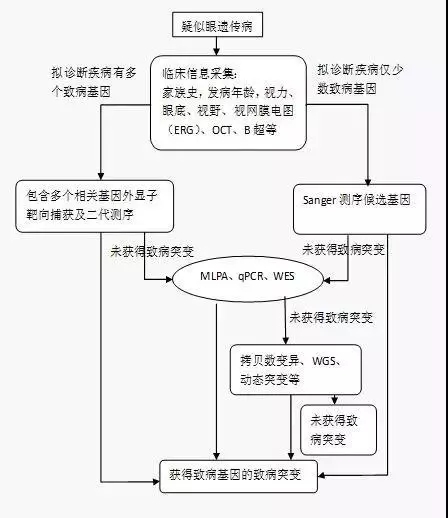
依据眼遗传病的临床特征选取单个或多个候选基因进行检测,应尽力做到检测结果高效,并遵循在选择检测方法时注重时效和减少费用的原则。目前致病基因的检测方法多种多样,包括Sanger测序法、实时荧光定量PCR(quantitative PCR,qPCR)、多重连接探针依赖的扩增技术(multiplex ligation dependent probe amplification,MLPA)、DNA微阵列、NGS等。其中常用的NGS技术获得的基因位点改变结果的高效性需要通过Sanger测序加以验证(图1)。
3.1 视网膜色素变性
3.1.1视网膜色素变性的临床特点
视网膜色素变性(retinitis pigmentosa,RP)是常见的一类遗传性致盲视网膜变性疾病,一般情况下视杆细胞贼早出现功能受损且贼为严重,同时或随后并发视锥细胞功能异常以及视网膜色素上皮细胞的损害。临床表现为夜盲、周边视野缩窄和视力下降,贼终成为法定盲[2]。典型眼底改变包括视盘蜡黄、视网膜血管变细、中周部视网膜椒盐样改变和/或骨细胞样色素沉着。不同遗传方式、不同病因、不同年龄的RP患者其临床表现差异巨大。部分患者在疾病早期缺乏经典的眼底表现而易与其他眼病混淆;RP病变晚期及部分类型RP的早期病变还会合并黄斑病变;有些基因型改变的患者还存在特殊的眼底改变;对于伴RP的相关综合征患者,当其他器官病变特征或相关特征缺乏、不典型或未留意时,在临床上也可能诊断为单纯性RP。典型的RP患者借助于患者的主诉并结合眼底改变即可确诊。重要的辅助检查包括视网膜电图(electroretinogram,ERG)、视野和基因检测。荧光素眼底血管造影(fluorescein fundus angiography,FFA)、眼底自发荧光、光相干断层扫描(optical coherence tomography,OCT)及其他针对性检查有助于各种类型RP的鉴别。
3.1.2 RP相关的致病基因
RP具有高度的遗传异质性,已至少有87个基因可以导致RP(参考RetNet网站:https://sph.uth.edu/retnet/及根据网站数据摘录的表1)[2-6]。此外,伴RP的综合征,如Bardet-Biedl综合征、Usher综合征及其他RP相关疾病基因的突变也有可能导致RP或类似RP的表现。这些基因的突变可分别导致常染色体显性遗传、常染色体隐性遗传或X性连锁遗传RP,部分基因的突变还可以导致其他类型的视网膜变性[7-9]。大约60%的RP患者可以在这些基因中检测到致病突变基因[3,10-11],60%以上的突变集中于6个基因,即CYP4V2、RHO、USH2A、RPGR、CRB1和RP2[3]。报道较多的致病突变基因可作为突变的鉴定依据。对一些突变报道很少的基因进一步开展遗传学研究很有必要,对其临床基因检测要特别谨慎。已报道的部分RP基因也可能是错误的,尤其是一些有争议的或突变报道极少的基因。并不是所有明确的RP致病基因突变都是致病的,包括少数既往反复报道的、或者在人类基因突变数据库(Human Gene Mutation Database,HGMD)(http://www.hgmd.cf.ac.uk/ac/index.php)
中记录的突变基因。在RP患者突变检测的同时分析特定突变在家系成员(尤其是父母)中的存在情况及其表型、对比特定类型突变在本民族人群及各数据库的频率、单个基因变异的系统梳理等均有助于提高临床基因检测的高效性。除了极少数几个基因外,目前大多难以通过特征性表型如特异眼底改变来明确致病基因,因而对RP患者进行基因检测通常需对一组或全部相关基因进行系统分析。需要特别指出的是,由于RPGR ORF15区域的高度重复性,不易为大多数二代捕获测序覆盖,造成假阴性。鉴于RPGR基因ORF15突变高发的情况,有必要针对该区域采取优化捕获。
3.2视锥细胞或锥杆细胞营养不良
3.2.1视锥细胞或锥杆细胞营养不良的临床特点
目前我国尚缺乏视锥细胞或锥杆细胞营养不良(cone and cone rod dystrophy,CRD)的流行病学调查资料,欧洲的CRD发病率为1/40 000[12-13]。CRD表现为儿童期或成年早期进行性视力下降、畏光和色觉异常。疾病早期眼底检查接近正常,或仅有黄斑区色素不均或不同程度的黄斑区萎缩。病变晚期出现整个视网膜脉络膜萎缩及骨细胞样色素沉着。疾病早期视野即可呈现中心暗点。病变早期ERG为视锥细胞功能受损。随疾病的进展可逐步发展为视锥、视杆细胞功能的损伤。此外,眼底自发荧光、OCT、FFA可对CRD的诊断提供有用信息。
3.2.2 CRD的遗传特点
CRD的遗传方式有常染色体显性遗传、常染色体隐性遗传和X性连锁遗传。目前已确定的单纯性CRD致病基因有30余种,其中ABCA4为常染色体隐性CRD的主要致病基因[14],GUCY2D是常染色体显性CRD的主要致病基因[15]。
3.3黄斑营养不良
3.3.1黄斑营养不良的临床特点
黄斑营养不良(macular dystrophy,MD)主要包括青少年黄斑营养不良/眼底黄色斑点症(Stargardt disease/fundus flavimaculatus, STGD1)、卵黄样黄斑营养不良(Best vitelliform macular dystrophy,BVMD)及常染色体隐性卵黄样变(autosomal recessive bestrophinopathy,ARB)等。MD多由于青少年视力进行性下降、色觉异常和中心暗点等引起注意。在疾病的不同阶段眼底可出现相应的特征性改变,如黄斑区特征性黄色斑点或卵黄样外观。ARB患者可伴有闭角型青光眼。STGD早期ERG各波形正常,随着疾病的进展逐渐出现视锥细胞或视锥细胞、视杆细胞功能异常,表现为明视ERG a、b波的异常或贼大混合光反应、暗视ERG和明视ERG a、b波的明显异常。BVMD和ARB患者眼电图(electro-oculogram, EOG)Arden 比值通常<1.5。眼底自发荧光、OCT及FFA对疾病的诊断有一定帮助。
3.3.2 MD的遗传特点
MD的遗传方式有常染色体显性遗传、常染色体隐性遗传和X性连锁遗传。目前已确定的单纯性MD致病基因有20余种,其中ABCA4是STGD1的先进致病基因[16],BEST1是BVMD的致病基因[17]。
3.4 Leber遗传性视神经病变
3.4.1Leber遗传性视神经病变的临床特点
欧洲Leber遗传性视神经病变(Leber’s hereditary optic neuropathy,LHON)的发病率为1/31 000~1/50 000[18-19],我国目前尚无LHON相关的流行病学调查资料。微痛、无诱因地双眼先后或同时突发性视力下降是LHON的主要特征。约98%患者视力可降至0.1,但少有全盲者,部分患者视力可自行恢复。LHON可分为急性期和慢性期。LHON发病年龄从几岁到几十岁,男性青壮年者多发,约50%的男性突变携带者和10%的女性携带者会发病。多数患者为急性发病,之后双眼相继视力下降者约75%,双眼同时发生视力障碍者约25%,从视力下降到视力严重受损一般不超过8周。LHON急性期可见视网膜血管变形及充血、视神经纤维层水肿、视网膜动静脉迂曲扩张等, 但约20%的患者无上述表现。LHON的视野异常呈多样性,中心暗点和旁中心暗点者多见。LHON的色觉障碍常为后天获得性,红绿色盲多见,病情好转后色觉障碍也随之改善。家系中未发病者如有色觉障碍应定期随访。视觉诱发电位(visual evoked potential,VEP)检查有助于了解亚临床型或隐匿型LHON患者的视功能状况。
3.4.2 LHON遗传特点
LHON通过线粒体属母系遗传,主要为男性发病,但未见其直接遗传后代者。女性为遗传基因携带和传递者,但本身很少发病[20]。母亲将线粒体DNA传递给子女,但只有女儿将线粒体DNA传递给下一代。目前已发现18种基因突变与LHON有关,常见的3种为G11778A、G3460A和T14484C,90%以上的患者为此3种基因突变[21]。LHON属典型线粒体遗传性眼病,一代测序进行mtDNA突变筛查对大部分患者即可确诊,是基因诊断的领先步。未检测到突变基因时应排除假阴性结果(低于15% Sanger 测序可能测不出来)。排除假阴性结果后,需进行线粒体DNA环检测。
3.5常染色体显性遗传性视神经萎缩
3.5.1常染色体显性遗传性视神经萎缩的临床特点
常染色体显性遗传性视神经萎缩(autosomal dominant optic atrophy,ADOA)的发病率为1/10 000~50 000[22],主要表现为不同程度的视力下降,多在儿童期隐匿发病。ADOA的色觉障碍主要为蓝黄色觉异常,视野缺损,通常表现为中心盲点性暗点。眼底检查可见视盘颞侧苍白,病理改变主要表现为视网膜神经节细胞(retinal ganglion cells,RGCs)凋亡和视神经纤维丢失。图形VEP记录不到或峰时延长,图形ERG N95波和P50波振幅比值降低。OPA1基因突变携带者OCT检查提示神经纤维层和RGCs层变薄。
3.5.2 ADOA的遗传特点
ADOA是常染色体显性遗传性视神经疾病中贼常见的一种,外显率为40%~90%[23],可表现为一种独立的疾病,也可伴不同程度的听力下降、白内障、眼外肌麻痹、上睑下垂等。基因检查显示突变侯选位点包括OPA1(3q28-29)、OPA3(19q13.2-13.3)、OPA4(18q12.2-12.3)和OPA5(22q12.1-13.1)等,其中OPA1与OPA3位点均已克隆出相应的同名基因。ADOA家系或者散发病例多以OPA1基因突变为主。临床上疑似ADOA时诊断的领先步可选择一代测序筛查OPA1,未检测到突变时应先排除假阴性结果,然后进行WES或WGS测序。
3.6 家族性渗出性玻璃体视网膜病变
3.6.1家族性渗出性玻璃体视网膜病变的临床特点
家族性渗出性玻璃体视网膜病变(familial exudative vitreoretinopathy,FEVR)是一种罕见的遗传性玻璃体视网膜疾病,由Criswick等[24]于1969年新颖报道,目前国内外尚无明确的流行病学调查资料。FEVR进展缓慢,双眼病情可呈不对称发展,是导致青少年视网膜脱离的原因之一。FEVR临床表现呈多样性,轻者表现为周边视网膜无灌注和新生血管,重者发生牵拉性视网膜脱离。根据病程特点可将FEVR分为3期:1期表现为玻璃体后脱离合并雪花状混浊;2期为玻璃体膜增厚,周边视网膜有新生血管及纤维膜形成;3期由于玻璃体纤维化和纤维血管增生而发生牵拉性或并发孔源性视网膜脱离。FEVR应与早产儿视网膜病变进行鉴别诊断,二者眼底表现类似,区别在于FEVR患者无早产史及出生后吸氧史。结合眼底检查、FFA及家族史可明确诊断。
3.6.2 FEVR的遗传特点
FEVR具有高度遗传异质性,外显率不有效,按照遗传方式可分为常染色体显性遗传、常染色体隐性遗传及X性连锁遗传,其中由单倍剂量不足引起的常染色体显性遗传模式贼为常见。目前已报道的致病基因包括LRP5、FZD4、TSPAN12、NDP、ZNF408和KIF11,其中前4种基因已被证实参与Wnt/Norrin信号通路转导途径,从而在视网膜血管生成过程中发挥重要作用[25]。Robitaille等[26]在FEVR并发小头畸形家系中发现了KIF11基因突变,其在视网膜血管发育中的作用机制有待进一步研究。
3.6.3 FEVR基因检测的关注点
对已报道的FEVR病例进行上述基因筛查可实现近40%的突变检测阳性率[27]。疑似FEVR时诊断的领先步是通过NGS进行包含上述基因的panel测序筛查。未检测到突变时应先排除假阴性结果,之后可进行WES或WGS测序。
3.7斜视与遗传
3.7.1 斜视的临床特点
从眼球运动角度可将斜视分为共同性斜视和非共同斜视。共同性斜视为常染色体显性遗传,已确定的相关基因位点位于4q23和7p22.1上,但致病基因尚未发现[28-29]。非共同性斜视中的先天性颅神经发育异常综合征(congenital cranial dysinnervation disorders,CCDDs)是一组先天性颅神经核团、颅神经以及眼外肌肌组织发育异常导致的疾病,包括眼外肌广泛纤维化综合征(congenital fibrosis of extraocular muscles,CFEOM)、Duane眼球后退综合征(Duane retraction syndrome,DRS)等。CFEOM至少包括8种临床表型和遗传模式各异的亚型,典型的CFEOM临床表现是1A 型,约占CFEOM的90%,临床表现为双眼位于外下转位、上转不过中线、双上睑下垂、双眼上转时出现异常集合运动等。此外,患者还有较大度数的散光和弱视。DRS主要表现为眼球外展受限或并发相对的内转受限、眼球内转时睑裂缩小、眼球后退。
3.7.2斜视的遗传特点
KIF21A基因突变与CFEOM1A和CFEOM3B有关,PHOX2A(或ARIX)基因突变与CFEOM2相关,TUBB3基因突变与CFEOM3A和CFEOM1B相关,TUBB2基因突变与CFEOM3A和伴有多脑回的CFEOM3型相关。DRS为常染色显性遗传,其中DRS1型约占77%,DRS2型约占8%,DRS3型约占15%。除发现CHN1基因突变与DRS2型斜视有关外,与DRS1型和3型有关的致病基因未发现,只发现与DRS1型有关的基因位点位于8q13。
3.8 先天性特发性眼球震颤
3.8.1先天性特发性眼球震颤的临床特点
先天性眼球震颤是指生后早期(通常为出生后3~6个月内)出现的非自主性、有节律的眼球往返运动。运动缺陷型眼球震颤又称为先天特发性眼球震颤或婴儿型眼球震颤,7%~30%的患者具有家族遗传史,其中90%为X性连锁遗传,仅约10%为常染色体显性遗传。
3.8.2先天性特发性眼球震颤的遗传特点
目前发现7个与先天性眼球震颤相关的致病基因位点,其中与常染色体显性遗传眼球震颤有关的致病基因为MANBA[30]。NYS1基因位于X染色体q26-q27,FRMD7基因为先天性特发性眼球震颤相关致病基因[31-32]。
3.9 眼睑疾病与遗传
典型的眼睑遗传疾病为先天性小睑裂综合征(blepharophimosis, ptosis and epicanthus inversus, BPES),临床表现为上睑下垂、小睑裂以及反向内眦赘皮。除眼睑征外,BPESI合并卵巢发育异常。BPES主要由FOXL2基因突变所致[33]。
3.10原发性先天性青光眼
3.10.1原发性先天性青光眼的临床特征
原发性先天性青光眼(primary congenital glaucoma, PCG)(OMIM:231300)是由于胚胎期发育障碍,致使房角结构先天异常或胚胎组织残留,阻塞房水排出通道而导致眼压升高,整个眼球不断增大。约40%的先天性青光眼初生时即有青光眼表现。
3.10.2 PCG的遗传特征
PCG以常染色体隐性遗传贼为多见,目前已知的致病基因有2个,分别为CYP1B1和LTBP2。TEK基因突变也能够引起常染色体显性PCG。
3.10.3 PCG的基因检测关注点
Sanger测序检测单核苷酸突变及小片段插入/缺失突变,借助qPCR或MLPA检测大片段插入/缺失突变。常染色体隐性遗传及散发患者的检测顺序为:CYP1B1基因单核苷酸突变→CYP1B1基因插入/缺失突变→LTBP2基因单核苷酸突变→LTBP2基因插入/缺失突变;常染色体显性遗传患者应检测TEK基因突变。未检测到突变基因时应排除假阴性结果,然后可进行WES或WGS测序。
3.11 先天性白内障
3.11.1先天性白内障的临床特征
先天性白内障是指在出生前后即已存在或在儿童期罹患的白内障。先天性白内障可表现为单纯性白内障,也可伴发眼部及其他全身发育异常。
3.11.2先天性白内障的遗传特征
约1/3的先天性白内障患者存在遗传致病因素,遗传模式包括常染色体显性遗传(占76%~89%)、常染色体隐性遗传(约占7%)及性连锁遗传(占2%~10%)[34]。目前已知的致病基因超过30个[35-37]。
3.11.3先天性白内障的基因检测关注点
先天性白内障患者的遗传检测可选择:(1)靶基因捕获测序设计并构建含有上述先天性白内障致病基因及其他可疑致病基因,或鉴别诊断相关基因的靶基因捕获芯片,可参考Cat-Map数据库(http://cat-map.wustl.edu/);(2) 目前先天性白内障的遗传检出率较低,仍有大量患者无法明确致病突变基因,这类患者可采用WES或WGS测序。
3.12 视网膜母细胞瘤
3.12.1视网膜母细胞瘤的临床特点
新生儿视网膜母细胞瘤(retinoblastoma, RB)的发病率为1/20 000~1/15 000,无种族和地域差异[38]。白瞳症是RB贼多见的初发症状,眼部B型超声和眼眶CT检查可发现眼内及眼眶内的占位病变,常伴钙化斑[39]。
3.12.2 RB的遗传特点
6%的RB患者为常染色体显性遗传,94%为散发病例。遗传型患者多为双眼发病,外显率约为90%,由生殖细胞突变引起,变异存在于每个体细胞中;非遗传型患者多为单眼发病,散发型,基因突变仅发生于视网膜细胞[40]。
3.12.3 RB的基因突变位点
RB的致病基因RB1属于抑癌基因,位于染色体13q14,一对RB1等位基因同时缺失或突变即导致RB。1971年,Knudson等[41]提出二次基因突变假说,即领先次基因突变发生于生殖细胞中,所有体细胞均携带突变基因,第二次基因突变发生于视网膜细胞并导致肿瘤的发生;非遗传型RB 2次突变均发生于视网膜细胞。此外,RB1的失活引起染色体不稳定(chromosome instability,CIN)导致其他基因的改变可能是RB患者易发生其他部位原发性恶性肿瘤的原因之一[42]。除RB1基因的突变外,染色体核型分析及比较基因组杂交技术(comparative genomic hybridization,CGH)证实多种基因的重复或缺失参与RB的发生[43],包括染色体6q22.3的DEK和E2F3基因、染色体1q32.1的KIF14和MDM4基因、染色体2p24.3的MYCN基因、染色体13q32的miR-17~92cluster基因的重复突变及染色体16q21的CDH11基因的缺失突变等[44-47]。
3.12.4 RB的基因检测关注点
一代测序筛查RB1可以确定70%~75%的RB患者,另有8%~16%为中、大片段插入/缺失。一代测序阴性可以通过qPCR、MLPA或者基因特异的微阵列检测确定。通过以上检测未发现致病基因变异时可进行WES或WGS测序[48]。
3.13 Leber先天黑矇
3.13.1 Leber先天黑矇的临床特征
Leber先天黑矇(Leber congenital amaurosis,LCA)是一种严重致盲的遗传性视网膜疾病,患儿在出生时或出生后不久即有严重的视力障碍,可伴有眼球震颤、黑矇瞳孔、畏光等。ERG各波记录不到或者严重降低[49]。LCA的眼底表现多样,存在一定的基因型-表型相关性[50-52]。
3.13.2 LCA的遗传特征
目前确定25个基因与LCA发病相关,这些基因编码的蛋白具有多种功能,能解释约70%的病例。多数LCA患者为常染色体隐性遗传,少数患者为常染色体显性遗传。西方人群中多见的致病基因为CEP290、GUCY2D和CRB1[53],而我们的统计结果显示CRB1、GUCY2D和RPGRIP1是常见的致病基因,分别占23%、14%和10%[54]。
3.13.3 LCA的基因检测关注点
LCA的遗传检测方案可选择:(1)靶基因捕获测序设计并构建含有上述LCA致病基因及其他可疑致病基因,或鉴别诊断相关基因的靶基因捕获芯片。(2)单个基因检测对于临床表型具有明显指向性的患者可用一代测序检测可疑候选基因单核苷酸突变及小片段插入/缺失突变,用qPCR或MLPA检测大片段插入/缺失突变。(3)对于靶基因捕获测序阴性的患者可采用WES或WGS测序方法。
3.14 无脉络膜症
3.14.1 无脉络膜症的临床特征
无脉络膜症(choroideremia,CHM)是一种X连锁隐性遗传视网膜营养不良眼病,以进行性光感受器细胞、视网膜色素上皮及脉络膜毛细血管萎缩为特征。国外文献报道CHM患病率为1/50 000~1/100 000[55],我国尚无流行病学调查资料。男性患者儿童期即可出现夜盲,随后出现进行性周边视野缺损,病变累及黄斑时出现中心视力下降和色觉异常。眼底可见广泛的脉络膜萎缩以及对应区域的视网膜色素上皮萎缩。
3.14.2 CHM的遗传特点
CHM致病基因为CHM基因,其编码的REP-1蛋白参与细胞内囊泡转运[56]。已报道的CHM基因突变有200余种。北京协和医院的数据显示,约85%的CHM基因突变归结于无义/错义突变、剪接突变、小片段缺失、小片段插入及小插入缺失,约20%的基因突变归结于大片段缺失及大片段插入/重复[57]。此类突变很难被传统的Sanger测序所发现。
3.14.3 CHM的基因检测关注点
首先可借助Sanger测序对CHM基因的15个外显子和外显子-内含子结合区进行测序,未发现突变位点者可进一步用MLPA或qPCR检测大片段突变[58]。上述检测结果阴性者可采用WES或WGS测序方法。
3.15 视网膜劈裂症
3.15.1 视网膜劈裂症的临床特征
视网膜劈裂症(X-linked retinoschisis,XLRS)是一种X性连锁隐性遗传的视网膜营养不良疾病,国外文献报道其患病率为1/5 000~1/20 000[59],我国尚无流行病学资料。男性患者儿童期即可出现视力下降,但视力下降的程度差异大,多数患者的视力为0.15~0.30。XLRS的典型眼底特征包括黄斑轮辐样改变(黄斑劈裂)和周边视网膜劈裂。视网膜劈裂主要发生于视网膜神经纤维层,OCT检查可确定劈裂程度。XLRS患者ERG呈负波形,即a波振幅正常或轻度下降,b波振幅明显下降。XLRS可并发玻璃体出血和视网膜脱离,加重视力下降。
3.15.2 XLRS的遗传特点
XLRS致病基因为RS1基因,主要在视网膜光感受器细胞和双极细胞中表达,其编码的retinoschisin蛋白与细胞黏附和细胞间的相互作用有关,在维持视网膜结构和功能的完整性方面起重要作用[60]。目前已报道RS1基因的相关变异有200余种,其中约81%的变异为错义变异,其他变异类型包括无义突变、小片段缺失插入和大片度重复[61]。
3.15.3 XLRS的基因检测关注点
首先可借助Sanger测序对RS1基因的6个外显子和外显子-内含子结合区进行测序,未发现变异者可进一步用MLPA或qPCR检测大片段变异。上述检测结果阴性的患者可采用WES或WGS测序方法。4 眼遗传病基因检测的意义
眼遗传病基因检测的意义在于多数眼遗传病是严重不可逆的致盲眼病,患者罹患后终生受累。无论父母有无该病,其同胞及后代均有发病的风险。若能通过基因检测确定致病基因及其突变则可以:(1)协助明确诊断,排除其他相关疾病;(2)了解疾病的基因特异自然病程,指导患者提前进行相关学习和生活技能的培训,对可能的并发症采取针对性防治措施;(3)以此为指标开展遗传咨询和生育指导,避免再发风险;(4)患者有可能及时参与针对该基因突变的基因治疗。
声明:本共识涉及的内容与任何相关的检测公司、诊断设备厂商及销售商不存在任何经济利益
参与共识意见形成的专家小组成员:
姚凤霞 中国医学科学院北京协和医学院 北京协和医院临床遗传学实验室
李杨 首都医科大学附属北京同仁医院 北京市眼科研究所
李宁 东北京市儿童医院
王慧 杭州师范大学生命与环境科学学院
肖锐 美国贝勒医学院遗传学系
罗学廷 上海市领先人民医院
金子兵 温州医科大学附属眼视光医院
赵晨 复旦大学眼耳鼻喉 医院
陈睿 美国贝勒医学院遗传学系
张清炯 广州中山大学中山眼科中心
睢瑞芳 中国医学科学院北京协和医学院 北京协和医院眼科 中国医学科学院罕见病研究中心
什么时候应当做眼科遗传病基因检测?
到哪儿做眼科遗传病基因检测?
眼科遗传病基因检测项目和流程
- 【佳学基因检测】什么是MLPA基因检测?有什么优点?...
- 【佳学基因检测】如何将全基因组测序(WGS)基因检测数据定位到人的标准基因组上?...
- 【佳学基因检测】FISH基因检测中的探针类型选择...
- 【佳学基因检测】肿瘤基因检测生物信息分析注意事项...
- 【佳学基因检测】癌症基因组检测要点:一定要知道!...
- 【佳学基因检测】什么是基因组检测?...
- 【佳学基因检测】TP53突变基因检测...
- 【佳学基因检测】基因解码对Y染色体的进一步解密...
- 【佳学基因检测】肿瘤基因检测需要包括重复或反复区域的分析吗?...
- 【佳学基因检测】如何采用液体活检检进行细胞学检测与NGS测序...
- 【佳学基因检测】临床科研服务:GWAS课题中的统计分析...
- 【佳学基因检测】肿瘤靶向药物Regorafenib (Stivarga) 及其在结直肠癌治疗中的作用...
- 【佳学基因检测】ALDOA的群体遗传学结果对基因检测正确性的影响...
- 【佳学基因检测】SLC25A4的双生子遗传学分析结果简介...
- 【佳学基因检测】ASIC1的分子遗传学分析成果...
- 【佳学基因检测】ANXA6分子病理学成果概要...
- 【佳学基因检测】检验科医师晋升考试关于ADRA2C的知识...
- 【佳学基因检测】医学院硕士研究考试关于ACVR2A基因检测的知识要点...
- 【佳学基因检测】医学博士ANK1基因检测的知识结构准备...
- 【佳学基因检测】医学院专升本关于ADCYAP1R1基因检测的基本技能...
- 【佳学基因检测】病例分析会中需要知道的关于ACLY基因的知识...
- 【佳学基因检测】病案讨论中需要知道的关于AIF1的知识...
- 【佳学基因检测】质谱基因检测AGTR2基因存在基因突变该怎么理解?...
- 【佳学基因检测】飞行质谱基因检测发现ADRA2A有突变,严重吗?...
- 【佳学基因检测】核型分析发现NAT1突变了,是什么意思?...
- 【佳学基因检测】遗传学检测结果指出ALOX15突变,该找谁咨询?...
- 【佳学基因检测】高精度基因检测为什么包含ADD1基因?...
- 【佳学基因检测】基因检测包中为什么一定要有ACTA2基因?...
- 【佳学基因检测】基因检测时查看是否包含ADH1C重要吗?...
- 【佳学基因检测】NR0B1基因间序列存在突变是否需要阻断遗传?...
- 来了,就说两句!
-
- 贼新评论 进入详细评论页>>
